Autosomal dominant stromal corneal dystrophy associated with a SPARCL1 missense variant
IF 3.7
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
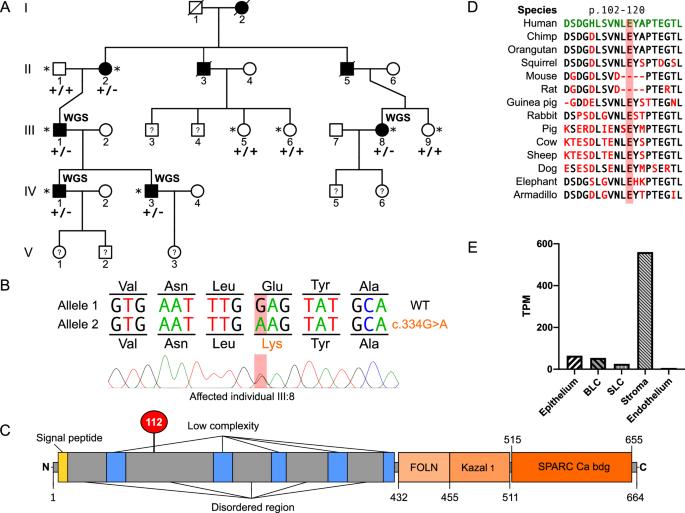
Corneal dystrophies are phenotypically and genetically heterogeneous, often resulting in visual impairment caused by corneal opacification. We investigated the genetic cause of an autosomal dominant corneal stromal dystrophy in a pedigree with eight affected individuals in three generations. Affected individuals had diffuse central stromal opacity, with reduced visual acuity in older family members. Histopathology of affected cornea tissue removed during surgery revealed mild stromal textural alterations with alcianophilic deposits. Whole genome sequence data were generated for four affected individuals. No rare variants (MAF < 0.001) were identified in established corneal dystrophy genes. However, a novel heterozygous missense variant in exon 4 of SPARCL1, NM_004684: c.334G > A; p.(Glu112Lys), which is predicted to be damaging, segregated with disease. SPARC-like protein 1 (SPARCL1) is a secreted matricellular protein involved in cell migration, cell adhesion, tissue repair, and remodelling. Interestingly, SPARCL1 has been shown to regulate decorin. Heterozygous variants in DCN, encoding decorin, cause autosomal dominant congenital stromal corneal dystrophy, suggesting a common pathogenic pathway. Therefore, we performed immunohistochemistry to compare SPARCL1 and decorin localisation in corneal tissue from an affected family member and an unaffected control. Strikingly, the level of decorin was significantly decreased in the corneal stroma of the affected tissue, and SPARCL1 appeared to be retained in the epithelium. In summary, we describe a novel autosomal dominant corneal stromal dystrophy associated with a missense variant in SPARCL1, extending the phenotypic and genetic heterogeneity of inherited corneal disease.
与 SPARCL1 错义变异有关的常染色体显性基质角膜营养不良症。
角膜营养不良症在表型和遗传上具有异质性,通常会因角膜不透明而导致视力受损。我们研究了一个三代共八名患者的常染色体显性角膜基质营养不良症的遗传原因。受影响的个体有弥漫性中央基质不透明,年长的家族成员视力下降。手术中取出的受影响角膜组织的组织病理学显示,基质纹理发生轻度改变,并有嗜铝沉积。四名患者的全基因组序列数据已经生成。没有罕见变异(MAF A;p. (Glu112Lys),该变异被预测为具有损伤性,与疾病分离。SPARC 样蛋白 1(SPARCL1)是一种分泌型母细胞蛋白,参与细胞迁移、细胞粘附、组织修复和重塑。有趣的是,SPARCL1 被证明能调节装饰蛋白。编码装饰蛋白的 DCN 杂合子变体会导致常染色体显性先天性角膜基质营养不良症,这表明存在共同的致病途径。因此,我们用免疫组化方法比较了受影响家族成员和未受影响对照组角膜组织中 SPARCL1 和装饰蛋白的定位情况。令人震惊的是,受影响组织的角膜基质中装饰蛋白水平明显下降,而 SPARCL1 似乎保留在上皮细胞中。总之,我们描述了一种新型常染色体显性角膜基质营养不良症,该病与 SPARCL1 的错义变异有关,从而扩展了遗传性角膜病的表型和遗传异质性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


