Daniel A. M. Pais, Jan-Peter A. Mayer, Karin Felderer, Maria B. Batalha, Timo Eichner, Sofia T. Santos, Raman Kumar, Sandra D. Silva, Hitto Kaufmann
{"title":"Holistic in silico developability assessment of novel classes of small proteins using publicly available sequence-based predictors","authors":"Daniel A. M. Pais, Jan-Peter A. Mayer, Karin Felderer, Maria B. Batalha, Timo Eichner, Sofia T. Santos, Raman Kumar, Sandra D. Silva, Hitto Kaufmann","doi":"10.1007/s10822-024-00569-x","DOIUrl":null,"url":null,"abstract":"<div><p>The development of novel therapeutic proteins is a lengthy and costly process, with an average attrition rate of 91% (Thomas et al. Clinical Development Success Rates and Contributing Factors 2011–2020, 2021). To increase the probability of success and ensure robust drug supply beyond approval, it is essential to assess the developability profile of new potential drug candidates as early and broadly as possible in development (Jain et al. MAbs, 2023. https://doi.org/10.1016/j.copbio.2011.06.002). Predicting these properties in silico is expected to be the next leap in innovation as it would enable significantly reduced development timelines combined with broader screens at lower costs. However, developing predictive algorithms typically requires substantial datasets generated under very defined conditions, a limiting factor especially for new classes of therapeutic proteins that hold immense clinical promise. Here we describe a strategy for assessing the developability of a novel class of small therapeutic Anticalin® proteins using machine learning in conjunction with a knowledge-driven approach. The knowledge-driven approach considers developability attributes such as aggregation propensity, charge variants, immunogenicity, specificity, thermal stability, hydrophobicity, and potential post-translational modifications, to calculate a holistic developability score. Based on sequence-derived descriptors as input parameters we established novel statistical models designed to predict the developability scores for Anticalin proteins. The best models yielded low root mean square errors across the entire dataset and were further validated by removing input data from individual screening campaigns and predicting developability scores for those drug candidates. The adoption of the described workflow will enable significantly streamlined preclinical development of Anticalin drug candidates and could potentially be applied to other therapeutic protein scaffolds.</p>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2024-08-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00569-x","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
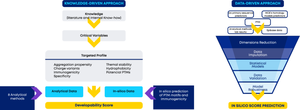
The development of novel therapeutic proteins is a lengthy and costly process, with an average attrition rate of 91% (Thomas et al. Clinical Development Success Rates and Contributing Factors 2011–2020, 2021). To increase the probability of success and ensure robust drug supply beyond approval, it is essential to assess the developability profile of new potential drug candidates as early and broadly as possible in development (Jain et al. MAbs, 2023. https://doi.org/10.1016/j.copbio.2011.06.002). Predicting these properties in silico is expected to be the next leap in innovation as it would enable significantly reduced development timelines combined with broader screens at lower costs. However, developing predictive algorithms typically requires substantial datasets generated under very defined conditions, a limiting factor especially for new classes of therapeutic proteins that hold immense clinical promise. Here we describe a strategy for assessing the developability of a novel class of small therapeutic Anticalin® proteins using machine learning in conjunction with a knowledge-driven approach. The knowledge-driven approach considers developability attributes such as aggregation propensity, charge variants, immunogenicity, specificity, thermal stability, hydrophobicity, and potential post-translational modifications, to calculate a holistic developability score. Based on sequence-derived descriptors as input parameters we established novel statistical models designed to predict the developability scores for Anticalin proteins. The best models yielded low root mean square errors across the entire dataset and were further validated by removing input data from individual screening campaigns and predicting developability scores for those drug candidates. The adoption of the described workflow will enable significantly streamlined preclinical development of Anticalin drug candidates and could potentially be applied to other therapeutic protein scaffolds.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


