Josiah Roberts, Biswas Rijal, Simon Divilov, Jon-Paul Maria, William G. Fahrenholtz, Douglas E. Wolfe, Donald W. Brenner, Stefano Curtarolo, Eva Zurek
{"title":"Machine learned interatomic potentials for ternary carbides trained on the AFLOW database","authors":"Josiah Roberts, Biswas Rijal, Simon Divilov, Jon-Paul Maria, William G. Fahrenholtz, Douglas E. Wolfe, Donald W. Brenner, Stefano Curtarolo, Eva Zurek","doi":"10.1038/s41524-024-01321-7","DOIUrl":null,"url":null,"abstract":"<p>Large-density functional theory (DFT) databases are a treasure trove of energies, forces, and stresses that can be used to train machine-learned interatomic potentials for atomistic modeling. Herein, we employ structural relaxations from the AFLOW database to train moment tensor potentials (MTPs) for four carbide systems: CHfTa, CHfZr, CMoW, and CTaTi. The resulting MTPs are used to relax ~6300 random symmetric structures, and are subsequently improved via active learning to generate robust potentials (RP) that can relax a wide variety of structures, and accurate potentials (AP) designed for the relaxation of low-energy systems. This protocol is shown to yield convex hulls that are indistinguishable from those predicted by AFLOW for the CHfTa, CHfZr, and CTaTi systems, and in the case of the CMoW system to predict thermodynamically stable structures that are not found within AFLOW, highlighting the potential of the employed protocol within crystal structure prediction. Relaxation of over three hundred (Mo<sub>1−<i>x</i></sub>W<sub><i>x</i></sub>)C stoichiometry crystals first with the RP then with the AP yields formation enthalpies that are in excellent agreement with those obtained via DFT.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"8 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01321-7","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
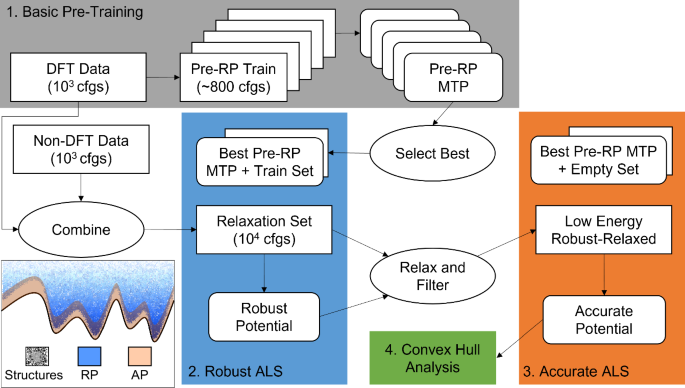
Large-density functional theory (DFT) databases are a treasure trove of energies, forces, and stresses that can be used to train machine-learned interatomic potentials for atomistic modeling. Herein, we employ structural relaxations from the AFLOW database to train moment tensor potentials (MTPs) for four carbide systems: CHfTa, CHfZr, CMoW, and CTaTi. The resulting MTPs are used to relax ~6300 random symmetric structures, and are subsequently improved via active learning to generate robust potentials (RP) that can relax a wide variety of structures, and accurate potentials (AP) designed for the relaxation of low-energy systems. This protocol is shown to yield convex hulls that are indistinguishable from those predicted by AFLOW for the CHfTa, CHfZr, and CTaTi systems, and in the case of the CMoW system to predict thermodynamically stable structures that are not found within AFLOW, highlighting the potential of the employed protocol within crystal structure prediction. Relaxation of over three hundred (Mo1−xWx)C stoichiometry crystals first with the RP then with the AP yields formation enthalpies that are in excellent agreement with those obtained via DFT.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


