Utilization of automated cilia analysis to characterize novel INPP5E variants in patients with non-syndromic retinitis pigmentosa
IF 3.7
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
INPP5E encodes inositol polyphosphate-5-phosphatase E, an enzyme involved in regulating the phosphatidylinositol (PIP) makeup of the primary cilium membrane. Pathogenic variants in INPP5E hence cause a variety of ciliopathies: genetic disorders caused by dysfunctional cilia. While the majority of these disorders are syndromic, such as the neuronal ciliopathy Joubert syndrome, in some cases patients will present with an isolated phenotype—most commonly non-syndromic retinitis pigmentosa (RP). Here, we report two novel variants in INPP5E identified in two patients with non-syndromic RP: patient 1 with compound heterozygous variants (c.1516C > T, p.(Q506*), and c.847G > A, p.(A283T)) and patient 2 with a homozygous variant (c.1073C > T, p.(P358L)). To determine whether these variants were causative for the phenotype in the patients, automated ciliary phenotyping of patient-derived dermal fibroblasts was performed for percent ciliation, cilium length, retrograde IFT trafficking, and INPP5E localization. In both patients, a decrease in ciliary length and loss of INPP5E localization in the primary cilia were seen. With these molecular findings, we can confirm functionally that the novel variants in INPP5E are causative for the RP phenotypes seen in both patients. Additionally, this study demonstrates the usefulness of utilizing ciliary phenotyping as an assistant in ciliopathy diagnosis and phenotyping.
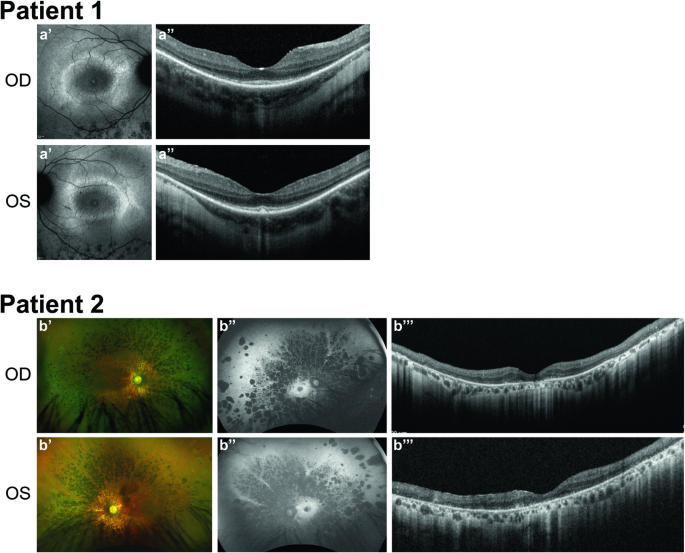
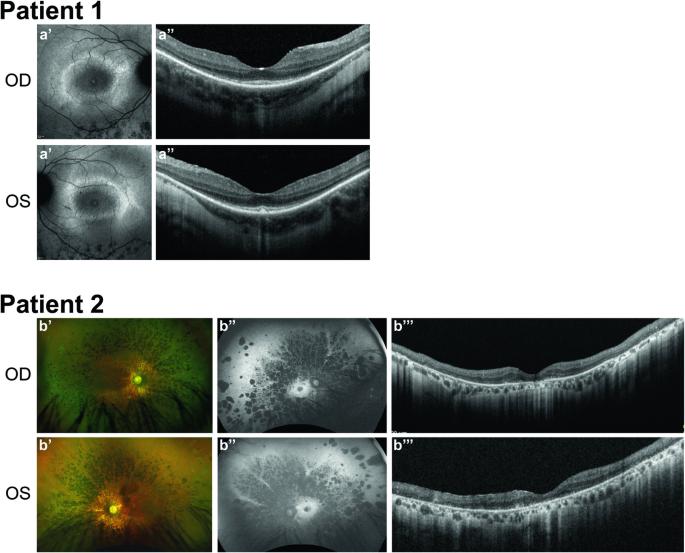
利用自动纤毛分析鉴定非综合征视网膜色素变性患者中的新型 INPP5E 变异。
INPP5E 编码肌醇多磷酸-5-磷酸酶 E,这是一种参与调节初级纤毛膜磷脂酰肌醇(PIP)构成的酶。因此,INPP5E 的致病变体会导致多种纤毛疾病:即由纤毛功能障碍引起的遗传性疾病。虽然这些疾病大多是综合征,如神经性纤毛病朱伯特综合征(neuronal ciliopathy Joubert syndrome),但在某些情况下,患者会表现出孤立的表型--最常见的是非综合征性色素性视网膜炎(retinitis pigmentosa,RP)。在此,我们报告了在两名非综合征 RP 患者中发现的 INPP5E 的两个新型变异:患者 1 患有复合杂合变异(c.1516C > T, p.(Q506*) 和 c.847G > A, p.(A283T)),患者 2 患有同源变异(c.1073C > T, p.(P358L))。为确定这些变异是否导致患者的表型,对患者来源的真皮成纤维细胞进行了自动纤毛表型分析,以确定纤毛对合率、纤毛长度、逆行 IFT 运输和 INPP5E 定位。两名患者的纤毛长度都有所下降,原发性纤毛中的 INPP5E 定位于缺失。通过这些分子研究结果,我们可以从功能上证实,INPP5E 的新型变体是导致这两名患者出现 RP 表型的原因。此外,这项研究还证明了利用纤毛表型作为纤毛病诊断和表型分析助手的实用性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

European Journal of Human Genetics
生物-生化与分子生物学
CiteScore
9.90
自引率
5.80%
发文量
216
审稿时长
2 months
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


