{"title":"De novo drug design as GPT language modeling: large chemistry models with supervised and reinforcement learning","authors":"Gavin Ye","doi":"10.1007/s10822-024-00559-z","DOIUrl":null,"url":null,"abstract":"<div><p>In recent years, generative machine learning algorithms have been successful in designing innovative drug-like molecules. SMILES is a sequence-like language used in most effective drug design models. Due to data’s sequential structure, models such as recurrent neural networks and transformers can design pharmacological compounds with optimized efficacy. Large language models have advanced recently, but their implications on drug design have not yet been explored. Although one study successfully pre-trained a <i>large chemistry model</i> (LCM), its application to specific tasks in drug discovery is unknown. In this study, the drug design task is modeled as a causal language modeling problem. Thus, the procedure of reward modeling, supervised fine-tuning, and proximal policy optimization was used to transfer the LCM to drug design, similar to Open AI’s ChatGPT and InstructGPT procedures. By combining the SMILES sequence with chemical descriptors, the novel efficacy evaluation model exceeded its performance compared to previous studies. After proximal policy optimization, the drug design model generated molecules with 99.2% having efficacy pIC<sub>50</sub> > 7 towards the amyloid precursor protein, with 100% of the generated molecules being valid and novel. This demonstrated the applicability of LCMs in drug discovery, with benefits including less data consumption while fine-tuning. The applicability of LCMs to drug discovery opens the door for larger studies involving reinforcement-learning with human feedback, where chemists provide feedback to LCMs and generate higher-quality molecules. LCMs’ ability to design similar molecules from datasets paves the way for more accessible, non-patented alternatives to drug molecules.</p></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2024-04-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-024-00559-z.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00559-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
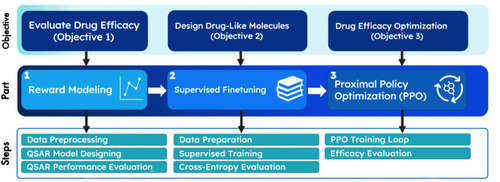
In recent years, generative machine learning algorithms have been successful in designing innovative drug-like molecules. SMILES is a sequence-like language used in most effective drug design models. Due to data’s sequential structure, models such as recurrent neural networks and transformers can design pharmacological compounds with optimized efficacy. Large language models have advanced recently, but their implications on drug design have not yet been explored. Although one study successfully pre-trained a large chemistry model (LCM), its application to specific tasks in drug discovery is unknown. In this study, the drug design task is modeled as a causal language modeling problem. Thus, the procedure of reward modeling, supervised fine-tuning, and proximal policy optimization was used to transfer the LCM to drug design, similar to Open AI’s ChatGPT and InstructGPT procedures. By combining the SMILES sequence with chemical descriptors, the novel efficacy evaluation model exceeded its performance compared to previous studies. After proximal policy optimization, the drug design model generated molecules with 99.2% having efficacy pIC50 > 7 towards the amyloid precursor protein, with 100% of the generated molecules being valid and novel. This demonstrated the applicability of LCMs in drug discovery, with benefits including less data consumption while fine-tuning. The applicability of LCMs to drug discovery opens the door for larger studies involving reinforcement-learning with human feedback, where chemists provide feedback to LCMs and generate higher-quality molecules. LCMs’ ability to design similar molecules from datasets paves the way for more accessible, non-patented alternatives to drug molecules.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


