Sophia M. N. Hönig, Florian Flachsenberg, Christiane Ehrt, Alexander Neumann, Robert Schmidt, Christian Lemmen, Matthias Rarey
{"title":"SpaceGrow: efficient shape-based virtual screening of billion-sized combinatorial fragment spaces","authors":"Sophia M. N. Hönig, Florian Flachsenberg, Christiane Ehrt, Alexander Neumann, Robert Schmidt, Christian Lemmen, Matthias Rarey","doi":"10.1007/s10822-024-00551-7","DOIUrl":null,"url":null,"abstract":"<p>The growing size of make-on-demand chemical libraries is posing new challenges to cheminformatics. These ultra-large chemical libraries became too large for exhaustive enumeration. Using a combinatorial approach instead, the resource requirement scales approximately with the number of synthons instead of the number of molecules. This gives access to billions or trillions of compounds as so-called chemical spaces with moderate hardware and in a reasonable time frame. While extremely performant ligand-based 2D methods exist in this context, 3D methods still largely rely on exhaustive enumeration and therefore fail to apply. Here, we present SpaceGrow: a novel shape-based 3D approach for ligand-based virtual screening of billions of compounds within hours on a single CPU. Compared to a conventional superposition tool, SpaceGrow shows comparable pose reproduction capacity based on RMSD and superior ranking performance while being orders of magnitude faster. Result assessment of two differently sized subsets of the eXplore space reveals a higher probability of finding superior results in larger spaces highlighting the potential of searching in ultra-large spaces. Furthermore, the application of SpaceGrow in a drug discovery workflow was investigated in four examples involving G protein-coupled receptors (GPCRs) with the aim to identify compounds with similar binding capabilities and molecular novelty.</p><p>SpaceGrow descriptor comparison for an example cut in the molecule of interest. Scoring scheme is implied for one fragment of this cut. </p>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2024-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10944417/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-024-00551-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
The growing size of make-on-demand chemical libraries is posing new challenges to cheminformatics. These ultra-large chemical libraries became too large for exhaustive enumeration. Using a combinatorial approach instead, the resource requirement scales approximately with the number of synthons instead of the number of molecules. This gives access to billions or trillions of compounds as so-called chemical spaces with moderate hardware and in a reasonable time frame. While extremely performant ligand-based 2D methods exist in this context, 3D methods still largely rely on exhaustive enumeration and therefore fail to apply. Here, we present SpaceGrow: a novel shape-based 3D approach for ligand-based virtual screening of billions of compounds within hours on a single CPU. Compared to a conventional superposition tool, SpaceGrow shows comparable pose reproduction capacity based on RMSD and superior ranking performance while being orders of magnitude faster. Result assessment of two differently sized subsets of the eXplore space reveals a higher probability of finding superior results in larger spaces highlighting the potential of searching in ultra-large spaces. Furthermore, the application of SpaceGrow in a drug discovery workflow was investigated in four examples involving G protein-coupled receptors (GPCRs) with the aim to identify compounds with similar binding capabilities and molecular novelty.
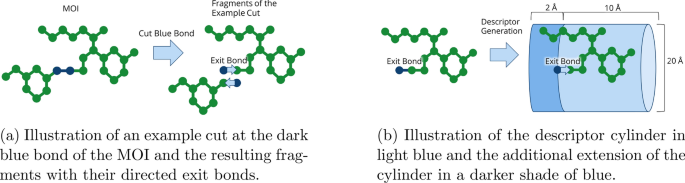
SpaceGrow descriptor comparison for an example cut in the molecule of interest. Scoring scheme is implied for one fragment of this cut.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


