Aleksei Kabedev, Christel A. S. Bergström, Per Larsson
{"title":"Molecular dynamics study on micelle-small molecule interactions: developing a strategy for an extensive comparison","authors":"Aleksei Kabedev, Christel A. S. Bergström, Per Larsson","doi":"10.1007/s10822-023-00541-1","DOIUrl":null,"url":null,"abstract":"<div><p>Theoretical predictions of the solubilizing capacity of micelles and vesicles present in intestinal fluid are important for the development of new delivery techniques and bioavailability improvement. A balance between accuracy and computational cost is a key factor for an extensive study of numerous compounds in diverse environments. In this study, we aimed to determine an optimal molecular dynamics (MD) protocol to evaluate small-molecule interactions with micelles composed of bile salts and phospholipids. MD simulations were used to produce free energy profiles for three drug molecules (danazol, probucol, and prednisolone) and one surfactant molecule (sodium caprate) as a function of the distance from the colloid center of mass. To address the challenges associated with such tasks, we compared different simulation setups, including freely assembled colloids versus pre-organized spherical micelles, full free energy profiles versus only a few points of interest, and a coarse-grained model versus an all-atom model. Our findings demonstrate that combining these techniques is advantageous for achieving optimal performance and accuracy when evaluating the solubilization capacity of micelles.</p><h3>Graphical abstract</h3><p>All-atom (AA) and coarse-grained (CG) umbrella sampling (US) simulations and point-wise free energy (FE) calculations were compared to their efficiency to computationally analyze the solubilization of active pharmaceutical ingredients in intestinal fluid colloids.</p>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":621,"journal":{"name":"Journal of Computer-Aided Molecular Design","volume":"38 1","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2023-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10822-023-00541-1.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computer-Aided Molecular Design","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10822-023-00541-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Theoretical predictions of the solubilizing capacity of micelles and vesicles present in intestinal fluid are important for the development of new delivery techniques and bioavailability improvement. A balance between accuracy and computational cost is a key factor for an extensive study of numerous compounds in diverse environments. In this study, we aimed to determine an optimal molecular dynamics (MD) protocol to evaluate small-molecule interactions with micelles composed of bile salts and phospholipids. MD simulations were used to produce free energy profiles for three drug molecules (danazol, probucol, and prednisolone) and one surfactant molecule (sodium caprate) as a function of the distance from the colloid center of mass. To address the challenges associated with such tasks, we compared different simulation setups, including freely assembled colloids versus pre-organized spherical micelles, full free energy profiles versus only a few points of interest, and a coarse-grained model versus an all-atom model. Our findings demonstrate that combining these techniques is advantageous for achieving optimal performance and accuracy when evaluating the solubilization capacity of micelles.
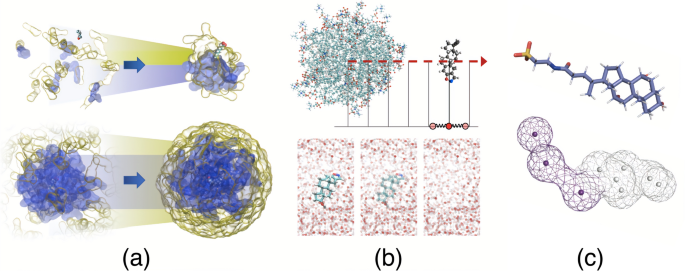
Graphical abstract
All-atom (AA) and coarse-grained (CG) umbrella sampling (US) simulations and point-wise free energy (FE) calculations were compared to their efficiency to computationally analyze the solubilization of active pharmaceutical ingredients in intestinal fluid colloids.
期刊介绍:
The Journal of Computer-Aided Molecular Design provides a form for disseminating information on both the theory and the application of computer-based methods in the analysis and design of molecules. The scope of the journal encompasses papers which report new and original research and applications in the following areas:
- theoretical chemistry;
- computational chemistry;
- computer and molecular graphics;
- molecular modeling;
- protein engineering;
- drug design;
- expert systems;
- general structure-property relationships;
- molecular dynamics;
- chemical database development and usage.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


