QM assisted ML for 19F NMR chemical shift prediction
Abstract
Background
Ligand-observed 19F NMR detection is an efficient method for screening libraries of fluorinated molecules in fragment-based drug design campaigns. Screening fluorinated molecules in large mixtures makes 19F NMR a high-throughput method. Typically, these mixtures are generated from pools of well-characterized fragments. By predicting 19F NMR chemical shift, mixtures could be generated for arbitrary fluorinated molecules facilitating for example focused screens.
Methods
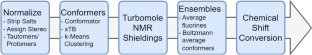
In a previous publication, we introduced a method to predict 19F NMR chemical shift using rooted fluorine fingerprints and machine learning (ML) methods. Having observed that the quality of the prediction depends on similarity to the training set, we here propose to assist the prediction with quantum mechanics (QM) based methods in cases where compounds are not well covered by a training set.
Results
Beyond similarity, the performance of ML methods could be associated with individual features in compounds. A combination of both could be used as a procedure to split input data sets into those that could be predicted by ML and those that required QM processing. We could show on a proprietary fluorinated fragment library, known as LEF (Local Environment of Fluorine), and a public Enamine data set of 19F NMR chemical shifts that ML and QM methods could synergize to outperform either method individually. Models built on Enamine data, as well as model building and QM workflow tools, can be found at https://github.com/PatrickPenner/lefshift and https://github.com/PatrickPenner/lefqm.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


