{"title":"Design and implementation of a hybrid cloud system for large-scale human genomic research.","authors":"Masao Nagasaki, Yayoi Sekiya, Akihiro Asakura, Ryo Teraoka, Ryoko Otokozawa, Hiroki Hashimoto, Takahisa Kawaguchi, Keiichiro Fukazawa, Yuichi Inadomi, Ken T Murata, Yasuyuki Ohkawa, Izumi Yamaguchi, Takamichi Mizuhara, Katsushi Tokunaga, Yuji Sekiya, Toshihiro Hanawa, Ryo Yamada, Fumihiko Matsuda","doi":"10.1038/s41439-023-00231-2","DOIUrl":null,"url":null,"abstract":"<p><p>In the field of genomic medical research, the amount of large-scale information continues to increase due to advances in measurement technologies, such as high-performance sequencing and spatial omics, as well as the progress made in genomic cohort studies involving more than one million individuals. Therefore, researchers require more computational resources to analyze this information. Here, we introduce a hybrid cloud system consisting of an on-premise supercomputer, science cloud, and public cloud at the Kyoto University Center for Genomic Medicine in Japan as a solution. This system can flexibly handle various heterogeneous computational resource-demanding bioinformatics tools while scaling the computational capacity. In the hybrid cloud system, we demonstrate the way to properly perform joint genotyping of whole-genome sequencing data for a large population of 11,238, which can be a bottleneck in sequencing data analysis. This system can be one of the reference implementations when dealing with large amounts of genomic medical data in research centers and organizations.</p>","PeriodicalId":36861,"journal":{"name":"Human Genome Variation","volume":"10 1","pages":"6"},"PeriodicalIF":1.0000,"publicationDate":"2023-02-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9908893/pdf/","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genome Variation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1038/s41439-023-00231-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 2
Abstract
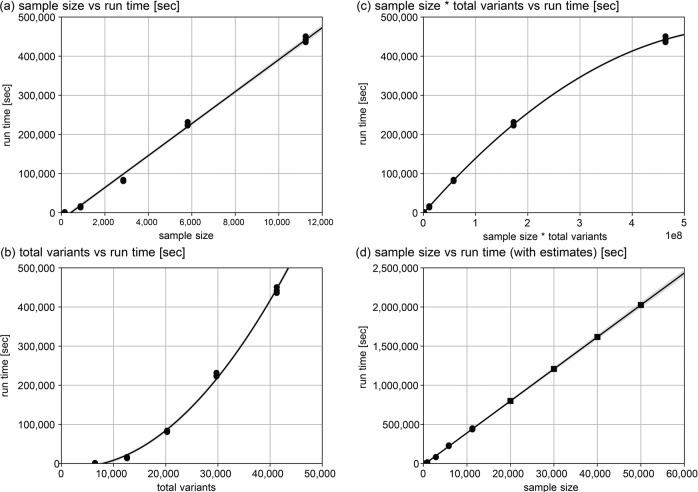
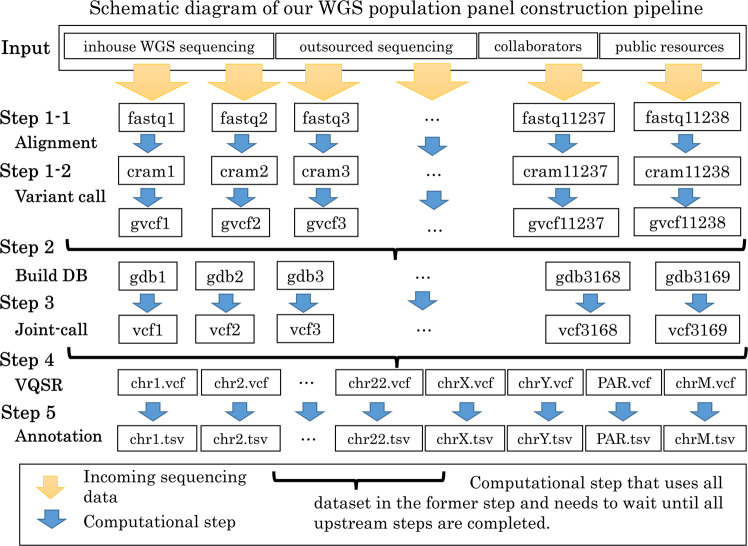
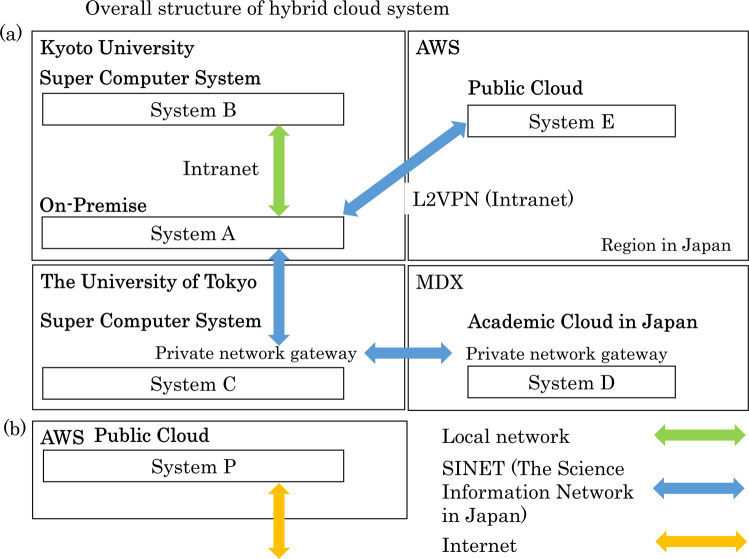
In the field of genomic medical research, the amount of large-scale information continues to increase due to advances in measurement technologies, such as high-performance sequencing and spatial omics, as well as the progress made in genomic cohort studies involving more than one million individuals. Therefore, researchers require more computational resources to analyze this information. Here, we introduce a hybrid cloud system consisting of an on-premise supercomputer, science cloud, and public cloud at the Kyoto University Center for Genomic Medicine in Japan as a solution. This system can flexibly handle various heterogeneous computational resource-demanding bioinformatics tools while scaling the computational capacity. In the hybrid cloud system, we demonstrate the way to properly perform joint genotyping of whole-genome sequencing data for a large population of 11,238, which can be a bottleneck in sequencing data analysis. This system can be one of the reference implementations when dealing with large amounts of genomic medical data in research centers and organizations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


