Sandeep Gurram, Vikram V Holla, Riyanka Kumari, Debjyoti Dhar, Nitish Kamble, Ravi Yadav, Babylakshmi Muthusamy, Pramod Kumar Pal
{"title":"Kufor-Rakeb综合征的强直性Opisthotinus:扩展表型和基因型谱。","authors":"Sandeep Gurram, Vikram V Holla, Riyanka Kumari, Debjyoti Dhar, Nitish Kamble, Ravi Yadav, Babylakshmi Muthusamy, Pramod Kumar Pal","doi":"10.14802/jmd.23098","DOIUrl":null,"url":null,"abstract":"Dear Editor, Kufor-Rakeb syndrome is a rare autosomal recessive disease, first described in 19941 in the Middle-Eastern country of Jordan. In 2006, biallelic mutations in the ATP13A2 gene were determined to be the underlying genetic etiology.2 More than 50 cases have been reported, including four cases from India.3,4 This syndrome is clinically characterized by juvenile-onset parkinsonism, supranuclear upgaze palsy, cognitive decline, pyramidal signs, visual hallucinations, oculogyric crisis, facial-faucialfinger mini myoclonus and dystonia in various combinations.5-7 In addition, biallelic loss-of-function mutations in the ATP13A2 gene can result in neuronal ceroid lipofuscinosis and complicated hereditary spastic paraplegia type 78 (SPG78).8,9 Here, we expand the phenotypic and genotypic spectrum of KuforRakeb syndrome by reporting dystonic opisthotonus in a patient with juvenile-onset parkinsonism and oculogyric crisis and a novel homozygous variant (NM_022089.4;c.705G>C) in the ATP13A2 gene. An 18-year-old man, born of a 3rd-degree consanguineous marriage (Figure 1), presented with episodes of uprolling of the eyeballs with retained awareness suggestive of oculogyric crisis, slurred speech, and drooling for 2 years and abnormal backward posturing of the trunk and neck while walking for 18 months. These symptoms occurred more frequently in the evenings and were reduced after a short nap. On examination, the patient had normal cognition, hypophonic speech, a reduced blink rate, mild upgaze impairment, and normal saccades and pursuits. He had right-side predominant asymmetrical parkinsonism with rigidity and bradykinesia but no tremor or postural instability. On walking, the patient had dystonic opisthotonus, which was less apparent when standing (Supplementary Video 1 in the online-only Data Supplement, Segment 1). In addition, the patient had hyperreflexia in the lower limbs with normal power and plantar response. The rest of the neurological and systemic examination results were normal. Routine blood investigation results, including hemogram, renal and liver function tests, serum electrolytes, copper and ceruloplasmin and magnetic resonance imaging (MRI) of the brain, were normal (Figure 1). Whole-exome sequencing revealed a novel homozygous missense variant in the ATP13A2 gene (NM_022089.4;c.705G>C;p.Glu235Asp). No other significant variant was found that could explain the clinical findings. The asymptomatic parents were found to be heterozygous carriers (Figure 1). The c.705G>C variant is novel and not reported in population databases. Computational prediction tools predicted that the p.Glu235Asp variant is likely to have functional consequences. According to Sorting Intolerant From Tolerant (SIFT), the variant was predicted to be deleterious (score = 0); PolyPhen-2 predicted it to be damaging (score = 0.983), and the Combined Annotation Dependent Depletion (CADD) score","PeriodicalId":16372,"journal":{"name":"Journal of Movement Disorders","volume":" ","pages":"343-346"},"PeriodicalIF":2.5000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/63/f7/jmd-23098.PMC10548071.pdf","citationCount":"0","resultStr":"{\"title\":\"Dystonic Opisthotonus in Kufor-Rakeb Syndrome: Expanding the Phenotypic and Genotypic Spectrum.\",\"authors\":\"Sandeep Gurram, Vikram V Holla, Riyanka Kumari, Debjyoti Dhar, Nitish Kamble, Ravi Yadav, Babylakshmi Muthusamy, Pramod Kumar Pal\",\"doi\":\"10.14802/jmd.23098\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Dear Editor, Kufor-Rakeb syndrome is a rare autosomal recessive disease, first described in 19941 in the Middle-Eastern country of Jordan. In 2006, biallelic mutations in the ATP13A2 gene were determined to be the underlying genetic etiology.2 More than 50 cases have been reported, including four cases from India.3,4 This syndrome is clinically characterized by juvenile-onset parkinsonism, supranuclear upgaze palsy, cognitive decline, pyramidal signs, visual hallucinations, oculogyric crisis, facial-faucialfinger mini myoclonus and dystonia in various combinations.5-7 In addition, biallelic loss-of-function mutations in the ATP13A2 gene can result in neuronal ceroid lipofuscinosis and complicated hereditary spastic paraplegia type 78 (SPG78).8,9 Here, we expand the phenotypic and genotypic spectrum of KuforRakeb syndrome by reporting dystonic opisthotonus in a patient with juvenile-onset parkinsonism and oculogyric crisis and a novel homozygous variant (NM_022089.4;c.705G>C) in the ATP13A2 gene. An 18-year-old man, born of a 3rd-degree consanguineous marriage (Figure 1), presented with episodes of uprolling of the eyeballs with retained awareness suggestive of oculogyric crisis, slurred speech, and drooling for 2 years and abnormal backward posturing of the trunk and neck while walking for 18 months. These symptoms occurred more frequently in the evenings and were reduced after a short nap. On examination, the patient had normal cognition, hypophonic speech, a reduced blink rate, mild upgaze impairment, and normal saccades and pursuits. He had right-side predominant asymmetrical parkinsonism with rigidity and bradykinesia but no tremor or postural instability. On walking, the patient had dystonic opisthotonus, which was less apparent when standing (Supplementary Video 1 in the online-only Data Supplement, Segment 1). In addition, the patient had hyperreflexia in the lower limbs with normal power and plantar response. The rest of the neurological and systemic examination results were normal. Routine blood investigation results, including hemogram, renal and liver function tests, serum electrolytes, copper and ceruloplasmin and magnetic resonance imaging (MRI) of the brain, were normal (Figure 1). Whole-exome sequencing revealed a novel homozygous missense variant in the ATP13A2 gene (NM_022089.4;c.705G>C;p.Glu235Asp). No other significant variant was found that could explain the clinical findings. The asymptomatic parents were found to be heterozygous carriers (Figure 1). The c.705G>C variant is novel and not reported in population databases. Computational prediction tools predicted that the p.Glu235Asp variant is likely to have functional consequences. According to Sorting Intolerant From Tolerant (SIFT), the variant was predicted to be deleterious (score = 0); PolyPhen-2 predicted it to be damaging (score = 0.983), and the Combined Annotation Dependent Depletion (CADD) score\",\"PeriodicalId\":16372,\"journal\":{\"name\":\"Journal of Movement Disorders\",\"volume\":\" \",\"pages\":\"343-346\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/63/f7/jmd-23098.PMC10548071.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Movement Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.14802/jmd.23098\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/7/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Movement Disorders","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.14802/jmd.23098","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Dystonic Opisthotonus in Kufor-Rakeb Syndrome: Expanding the Phenotypic and Genotypic Spectrum.
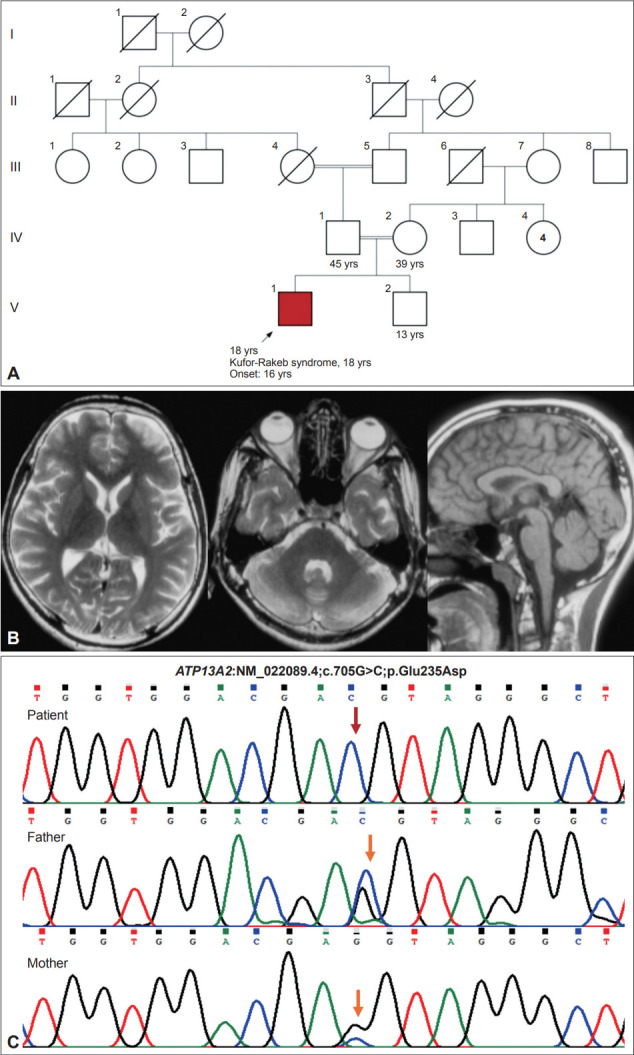
Dear Editor, Kufor-Rakeb syndrome is a rare autosomal recessive disease, first described in 19941 in the Middle-Eastern country of Jordan. In 2006, biallelic mutations in the ATP13A2 gene were determined to be the underlying genetic etiology.2 More than 50 cases have been reported, including four cases from India.3,4 This syndrome is clinically characterized by juvenile-onset parkinsonism, supranuclear upgaze palsy, cognitive decline, pyramidal signs, visual hallucinations, oculogyric crisis, facial-faucialfinger mini myoclonus and dystonia in various combinations.5-7 In addition, biallelic loss-of-function mutations in the ATP13A2 gene can result in neuronal ceroid lipofuscinosis and complicated hereditary spastic paraplegia type 78 (SPG78).8,9 Here, we expand the phenotypic and genotypic spectrum of KuforRakeb syndrome by reporting dystonic opisthotonus in a patient with juvenile-onset parkinsonism and oculogyric crisis and a novel homozygous variant (NM_022089.4;c.705G>C) in the ATP13A2 gene. An 18-year-old man, born of a 3rd-degree consanguineous marriage (Figure 1), presented with episodes of uprolling of the eyeballs with retained awareness suggestive of oculogyric crisis, slurred speech, and drooling for 2 years and abnormal backward posturing of the trunk and neck while walking for 18 months. These symptoms occurred more frequently in the evenings and were reduced after a short nap. On examination, the patient had normal cognition, hypophonic speech, a reduced blink rate, mild upgaze impairment, and normal saccades and pursuits. He had right-side predominant asymmetrical parkinsonism with rigidity and bradykinesia but no tremor or postural instability. On walking, the patient had dystonic opisthotonus, which was less apparent when standing (Supplementary Video 1 in the online-only Data Supplement, Segment 1). In addition, the patient had hyperreflexia in the lower limbs with normal power and plantar response. The rest of the neurological and systemic examination results were normal. Routine blood investigation results, including hemogram, renal and liver function tests, serum electrolytes, copper and ceruloplasmin and magnetic resonance imaging (MRI) of the brain, were normal (Figure 1). Whole-exome sequencing revealed a novel homozygous missense variant in the ATP13A2 gene (NM_022089.4;c.705G>C;p.Glu235Asp). No other significant variant was found that could explain the clinical findings. The asymptomatic parents were found to be heterozygous carriers (Figure 1). The c.705G>C variant is novel and not reported in population databases. Computational prediction tools predicted that the p.Glu235Asp variant is likely to have functional consequences. According to Sorting Intolerant From Tolerant (SIFT), the variant was predicted to be deleterious (score = 0); PolyPhen-2 predicted it to be damaging (score = 0.983), and the Combined Annotation Dependent Depletion (CADD) score

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


