{"title":"乳腺癌启动子甲基化调控的差异表达基因","authors":"Samar Sindi, Norah Hamdi, Sabah Hassan, Magdah Ganash, Mona Alharbi, Najla Alburae, Sheren Azhari, Shadi Alkhayyat, Ayman Linjawi, Heba Alkhatabi, Aisha Elaimi, Ghadeer Alrefaei, Nouf Alsubhi, Aziza Alrafiah, Safiah Alhazmi","doi":"10.2147/BCTT.S408711","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Breast cancer is one of the most common malignancies among women. Recent studies revealed that differentially methylated regions (DMRs) are implicated in regulating gene expression. The goal of this research was to determine which genes and pathways are dysregulated in breast cancer when their promoters are methylated in an abnormal way, leading to differential expression. Whole-genome bisulfite sequencing was applied to analyze DMRs for eight peripheral blood samples collected from five Saudi females diagnosed with stages I and II of breast cancer aligned with three normal females. Three of those patients and three normal samples were used to determine differentially expressed genes (DEG) using Illumina platform NovaSeq PE150.</p><p><strong>Results: </strong>Based on ontology (GO) and KEGG pathways, the analysis indicated that DMGs and DEG are closely related to associated processes, such as ubiquitin-protein transferase activity, ubiquitin-mediated proteolysis, and oxidative phosphorylation. The findings indicated a potentially significant association between global hypomethylation and breast cancer in Saudi patients. Our results revealed 81 differentially promoter-methylated and expressed genes. The most significant differentially methylated and expressed genes found in gene ontology (GO) are pumilio RNA binding family member 1 (<i>PUM1</i>) and zinc finger AN1-type containing 2B (<i>ZFAND2B</i>) also known as (<i>AIRAPL</i>).</p><p><strong>Conclusion: </strong>The essential outcomes of this study suggested that aberrant hypermethylation at crucial genes that have significant parts in the molecular pathways of breast cancer could be used as a potential prognostic biomarker for breast cancer.</p>","PeriodicalId":9106,"journal":{"name":"Breast Cancer : Targets and Therapy","volume":null,"pages":null},"PeriodicalIF":3.3000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/fa/e0/bctt-15-435.PMC10332364.pdf","citationCount":"0","resultStr":"{\"title\":\"Promoter Methylation-Regulated Differentially Expressed Genes in Breast Cancer.\",\"authors\":\"Samar Sindi, Norah Hamdi, Sabah Hassan, Magdah Ganash, Mona Alharbi, Najla Alburae, Sheren Azhari, Shadi Alkhayyat, Ayman Linjawi, Heba Alkhatabi, Aisha Elaimi, Ghadeer Alrefaei, Nouf Alsubhi, Aziza Alrafiah, Safiah Alhazmi\",\"doi\":\"10.2147/BCTT.S408711\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Breast cancer is one of the most common malignancies among women. Recent studies revealed that differentially methylated regions (DMRs) are implicated in regulating gene expression. The goal of this research was to determine which genes and pathways are dysregulated in breast cancer when their promoters are methylated in an abnormal way, leading to differential expression. Whole-genome bisulfite sequencing was applied to analyze DMRs for eight peripheral blood samples collected from five Saudi females diagnosed with stages I and II of breast cancer aligned with three normal females. Three of those patients and three normal samples were used to determine differentially expressed genes (DEG) using Illumina platform NovaSeq PE150.</p><p><strong>Results: </strong>Based on ontology (GO) and KEGG pathways, the analysis indicated that DMGs and DEG are closely related to associated processes, such as ubiquitin-protein transferase activity, ubiquitin-mediated proteolysis, and oxidative phosphorylation. The findings indicated a potentially significant association between global hypomethylation and breast cancer in Saudi patients. Our results revealed 81 differentially promoter-methylated and expressed genes. The most significant differentially methylated and expressed genes found in gene ontology (GO) are pumilio RNA binding family member 1 (<i>PUM1</i>) and zinc finger AN1-type containing 2B (<i>ZFAND2B</i>) also known as (<i>AIRAPL</i>).</p><p><strong>Conclusion: </strong>The essential outcomes of this study suggested that aberrant hypermethylation at crucial genes that have significant parts in the molecular pathways of breast cancer could be used as a potential prognostic biomarker for breast cancer.</p>\",\"PeriodicalId\":9106,\"journal\":{\"name\":\"Breast Cancer : Targets and Therapy\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/fa/e0/bctt-15-435.PMC10332364.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Breast Cancer : Targets and Therapy\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2147/BCTT.S408711\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Breast Cancer : Targets and Therapy","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2147/BCTT.S408711","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ONCOLOGY","Score":null,"Total":0}
Promoter Methylation-Regulated Differentially Expressed Genes in Breast Cancer.
Background: Breast cancer is one of the most common malignancies among women. Recent studies revealed that differentially methylated regions (DMRs) are implicated in regulating gene expression. The goal of this research was to determine which genes and pathways are dysregulated in breast cancer when their promoters are methylated in an abnormal way, leading to differential expression. Whole-genome bisulfite sequencing was applied to analyze DMRs for eight peripheral blood samples collected from five Saudi females diagnosed with stages I and II of breast cancer aligned with three normal females. Three of those patients and three normal samples were used to determine differentially expressed genes (DEG) using Illumina platform NovaSeq PE150.
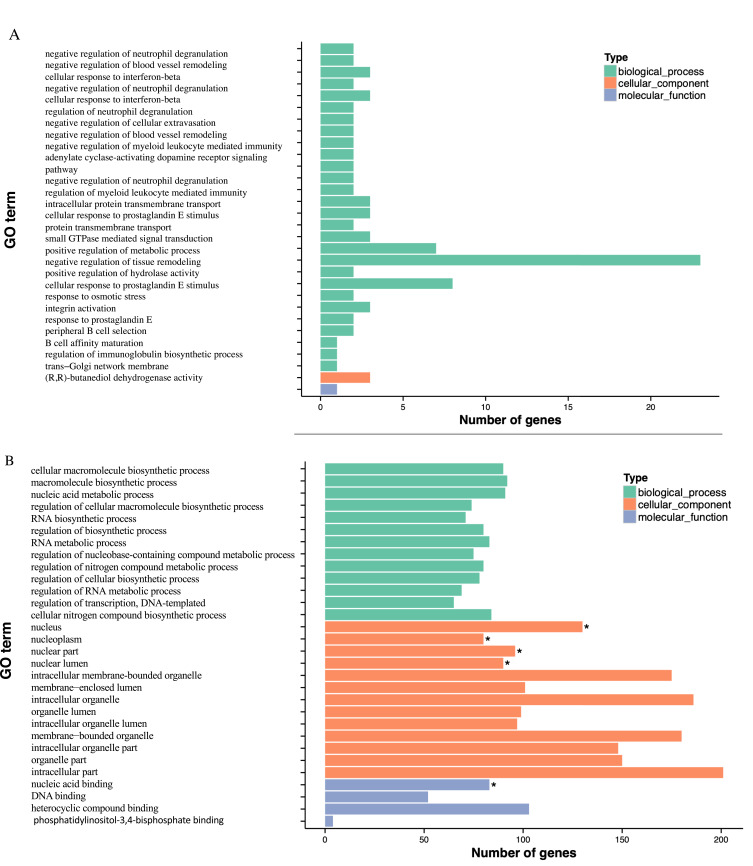
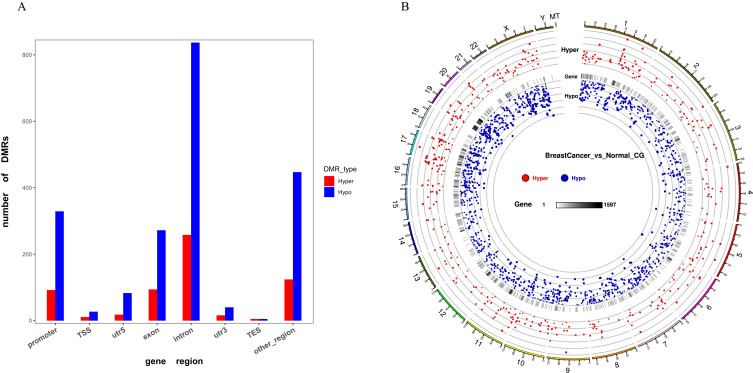
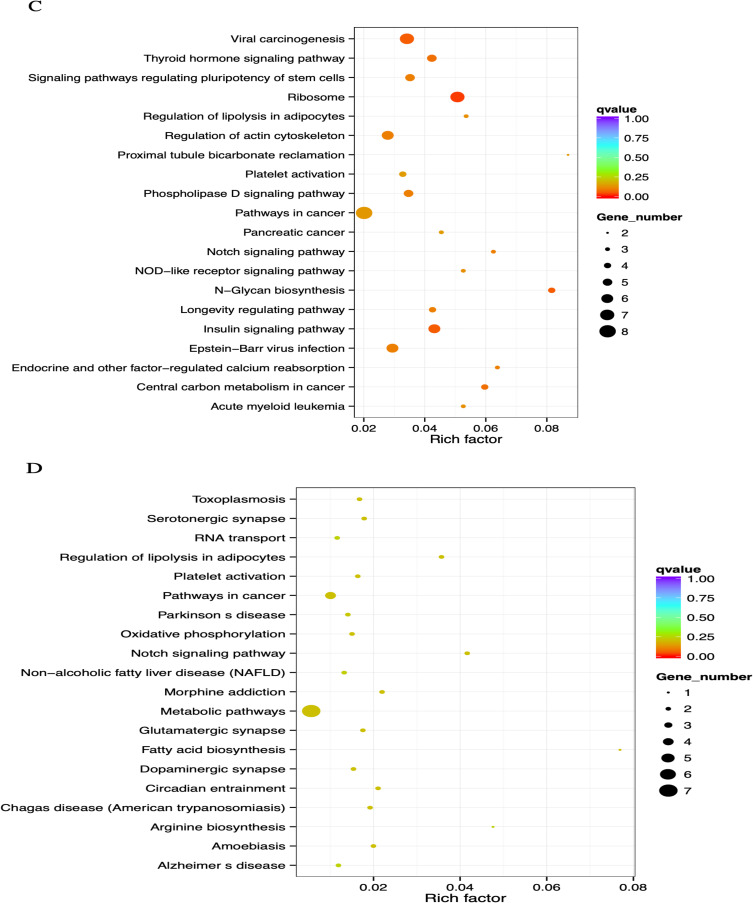
Results: Based on ontology (GO) and KEGG pathways, the analysis indicated that DMGs and DEG are closely related to associated processes, such as ubiquitin-protein transferase activity, ubiquitin-mediated proteolysis, and oxidative phosphorylation. The findings indicated a potentially significant association between global hypomethylation and breast cancer in Saudi patients. Our results revealed 81 differentially promoter-methylated and expressed genes. The most significant differentially methylated and expressed genes found in gene ontology (GO) are pumilio RNA binding family member 1 (PUM1) and zinc finger AN1-type containing 2B (ZFAND2B) also known as (AIRAPL).
Conclusion: The essential outcomes of this study suggested that aberrant hypermethylation at crucial genes that have significant parts in the molecular pathways of breast cancer could be used as a potential prognostic biomarker for breast cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


