Ja Hye Kim, Gu-Hwan Kim, Han-Wook Yoo, Jin-Ho Choi
{"title":"诊断21-羟化酶缺乏症的分子基础和基因检测策略,包括CAH-X综合征。","authors":"Ja Hye Kim, Gu-Hwan Kim, Han-Wook Yoo, Jin-Ho Choi","doi":"10.6065/apem.2346108.054","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital adrenal hyperplasia (CAH) is a group of autosomally recessive disorders that result from impaired synthesis of glucocorticoid and mineralocorticoid. Most cases (~95%) are caused by mutations in the CYP21A2 gene, which encodes steroid 21-hydroxylase. CAH patients manifest a wide phenotypic spectrum according to their degree of residual enzyme activity. CYP21A2 and its pseudogene (CYP21A1P) are located 30 kb apart in the 6q21.3 region and share approximately 98% of their sequences in the coding region. Both genes are aligned in tandem with the C4, SKT19, and TNX genes, forming 2 segments of the RCCX modules that are arranged as STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB. The high sequence homology between the active gene and pseudogene leads to frequent microconversions and large rearrangements through intergenic recombination. The TNXB gene encodes an extracellular matrix glycoprotein, tenascin-X (TNX), and defects in TNXB cause Ehlers-Danlos syndrome. Deletions affecting both CYP21A2 and TNXB result in a contiguous gene deletion syndrome known as CAH-X syndrome. Because of the high homology between CYP21A2 and CYP21A1P, genetic testing for CAH should include an evaluation of copy number variations, as well as Sanger sequencing. Although it poses challenges for genetic testing, a large number of mutations and their associated phenotypes have been identified, which has helped to establish genotype-phenotype correlations. The genotype is helpful for guiding early treatment, predicting the clinical phenotype and prognosis, and providing genetic counseling. In particular, it can help ensure proper management of the potential complications of CAH-X syndrome, such as musculoskeletal and cardiac defects. This review focuses on the molecular pathophysiology and genetic diagnosis of 21-hydroxylase deficiency and highlights genetic testing strategies for CAH-X syndrome.</p>","PeriodicalId":44915,"journal":{"name":"Annals of Pediatric Endocrinology & Metabolism","volume":null,"pages":null},"PeriodicalIF":2.8000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f4/b9/apem-2346108-054.PMC10329939.pdf","citationCount":"1","resultStr":"{\"title\":\"Molecular basis and genetic testing strategies for diagnosing 21-hydroxylase deficiency, including CAH-X syndrome.\",\"authors\":\"Ja Hye Kim, Gu-Hwan Kim, Han-Wook Yoo, Jin-Ho Choi\",\"doi\":\"10.6065/apem.2346108.054\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Congenital adrenal hyperplasia (CAH) is a group of autosomally recessive disorders that result from impaired synthesis of glucocorticoid and mineralocorticoid. Most cases (~95%) are caused by mutations in the CYP21A2 gene, which encodes steroid 21-hydroxylase. CAH patients manifest a wide phenotypic spectrum according to their degree of residual enzyme activity. CYP21A2 and its pseudogene (CYP21A1P) are located 30 kb apart in the 6q21.3 region and share approximately 98% of their sequences in the coding region. Both genes are aligned in tandem with the C4, SKT19, and TNX genes, forming 2 segments of the RCCX modules that are arranged as STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB. The high sequence homology between the active gene and pseudogene leads to frequent microconversions and large rearrangements through intergenic recombination. The TNXB gene encodes an extracellular matrix glycoprotein, tenascin-X (TNX), and defects in TNXB cause Ehlers-Danlos syndrome. Deletions affecting both CYP21A2 and TNXB result in a contiguous gene deletion syndrome known as CAH-X syndrome. Because of the high homology between CYP21A2 and CYP21A1P, genetic testing for CAH should include an evaluation of copy number variations, as well as Sanger sequencing. Although it poses challenges for genetic testing, a large number of mutations and their associated phenotypes have been identified, which has helped to establish genotype-phenotype correlations. The genotype is helpful for guiding early treatment, predicting the clinical phenotype and prognosis, and providing genetic counseling. In particular, it can help ensure proper management of the potential complications of CAH-X syndrome, such as musculoskeletal and cardiac defects. This review focuses on the molecular pathophysiology and genetic diagnosis of 21-hydroxylase deficiency and highlights genetic testing strategies for CAH-X syndrome.</p>\",\"PeriodicalId\":44915,\"journal\":{\"name\":\"Annals of Pediatric Endocrinology & Metabolism\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f4/b9/apem-2346108-054.PMC10329939.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Pediatric Endocrinology & Metabolism\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.6065/apem.2346108.054\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Pediatric Endocrinology & Metabolism","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.6065/apem.2346108.054","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Molecular basis and genetic testing strategies for diagnosing 21-hydroxylase deficiency, including CAH-X syndrome.
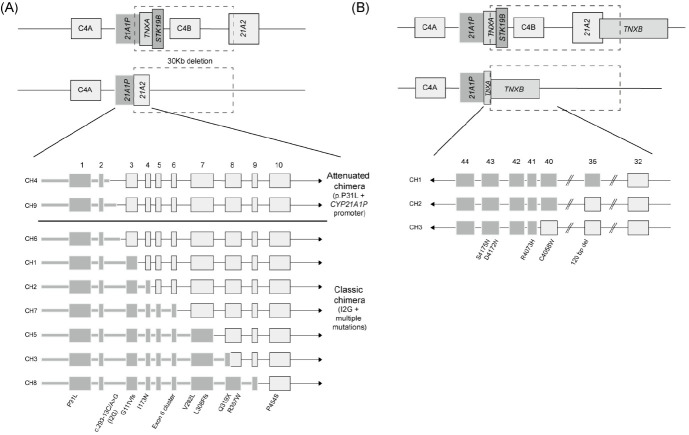
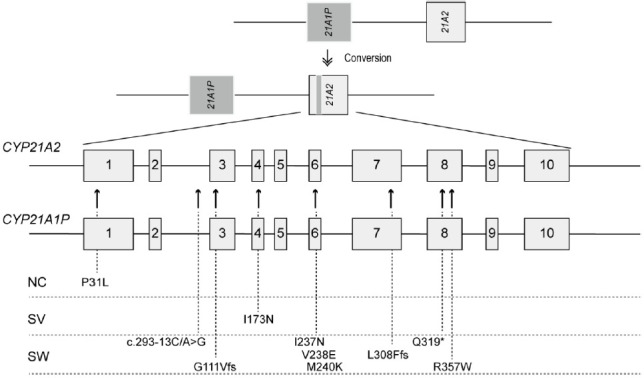
Congenital adrenal hyperplasia (CAH) is a group of autosomally recessive disorders that result from impaired synthesis of glucocorticoid and mineralocorticoid. Most cases (~95%) are caused by mutations in the CYP21A2 gene, which encodes steroid 21-hydroxylase. CAH patients manifest a wide phenotypic spectrum according to their degree of residual enzyme activity. CYP21A2 and its pseudogene (CYP21A1P) are located 30 kb apart in the 6q21.3 region and share approximately 98% of their sequences in the coding region. Both genes are aligned in tandem with the C4, SKT19, and TNX genes, forming 2 segments of the RCCX modules that are arranged as STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB. The high sequence homology between the active gene and pseudogene leads to frequent microconversions and large rearrangements through intergenic recombination. The TNXB gene encodes an extracellular matrix glycoprotein, tenascin-X (TNX), and defects in TNXB cause Ehlers-Danlos syndrome. Deletions affecting both CYP21A2 and TNXB result in a contiguous gene deletion syndrome known as CAH-X syndrome. Because of the high homology between CYP21A2 and CYP21A1P, genetic testing for CAH should include an evaluation of copy number variations, as well as Sanger sequencing. Although it poses challenges for genetic testing, a large number of mutations and their associated phenotypes have been identified, which has helped to establish genotype-phenotype correlations. The genotype is helpful for guiding early treatment, predicting the clinical phenotype and prognosis, and providing genetic counseling. In particular, it can help ensure proper management of the potential complications of CAH-X syndrome, such as musculoskeletal and cardiac defects. This review focuses on the molecular pathophysiology and genetic diagnosis of 21-hydroxylase deficiency and highlights genetic testing strategies for CAH-X syndrome.
期刊介绍:
The Annals of Pediatric Endocrinology & Metabolism Journal is the official publication of the Korean Society of Pediatric Endocrinology. Its formal abbreviated title is “Ann Pediatr Endocrinol Metab”. It is a peer-reviewed open access journal of medicine published in English. The journal was launched in 1996 under the title of ‘Journal of Korean Society of Pediatric Endocrinology’ until 2011 (pISSN 1226-2242). Since 2012, the title is now changed to ‘Annals of Pediatric Endocrinology & Metabolism’. The Journal is published four times per year on the last day of March, June, September, and December. It is widely distributed for free to members of the Korean Society of Pediatric Endocrinology, medical schools, libraries, and academic institutions. The journal is indexed/tracked/covered by web sites of PubMed Central, PubMed, Emerging Sources Citation Index (ESCI), Scopus, EBSCO, EMBASE, KoreaMed, KoMCI, KCI, Science Central, DOI/CrossRef, Directory of Open Access Journals(DOAJ), and Google Scholar. The aims of Annals of Pediatric Endocrinology & Metabolism are to contribute to the advancements in the fields of pediatric endocrinology & metabolism through the scientific reviews and interchange of all of pediatric endocrinology and metabolism. It aims to reflect the latest clinical, translational, and basic research trends from worldwide valuable achievements. In addition, genome research, epidemiology, public education and clinical practice guidelines in each country are welcomed for publication. The Journal particularly focuses on research conducted with Asian-Pacific children whose genetic and environmental backgrounds are different from those of the Western. Area of specific interest include the following : Growth, puberty, glucose metabolism including diabetes mellitus, obesity, nutrition, disorders of sexual development, pituitary, thyroid, parathyroid, adrenal cortex, bone or other endocrine and metabolic disorders from infancy through adolescence.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


