Ian C Smith, Chantal A Pileggi, Ying Wang, Kristin Kernohan, Taila Hartley, Hugh J McMillan, Marcos Loreto Sampaio, Gerd Melkus, John Woulfe, Gaganvir Parmar, Pierre R Bourque, Ari Breiner, Jocelyn Zwicker, C Elizabeth Pringle, Olga Jarinova, Hanns Lochmüller, David A Dyment, Bernard Brais, Kym M Boycott, Siegfried Hekimi, Mary-Ellen Harper, Jodi Warman-Chardon
{"title":"遗传性运动神经病兄弟姐妹中COQ7的新纯合变异。","authors":"Ian C Smith, Chantal A Pileggi, Ying Wang, Kristin Kernohan, Taila Hartley, Hugh J McMillan, Marcos Loreto Sampaio, Gerd Melkus, John Woulfe, Gaganvir Parmar, Pierre R Bourque, Ari Breiner, Jocelyn Zwicker, C Elizabeth Pringle, Olga Jarinova, Hanns Lochmüller, David A Dyment, Bernard Brais, Kym M Boycott, Siegfried Hekimi, Mary-Ellen Harper, Jodi Warman-Chardon","doi":"10.1212/NXG.0000000000200048","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Coenzyme Q<sub>10</sub> (CoQ<sub>10</sub>) is an important electron carrier and antioxidant. The COQ7 enzyme catalyzes the hydroxylation of 5-demethoxyubiquinone-10 (DMQ<sub>10</sub>), the second-to-last step in the CoQ<sub>10</sub> biosynthesis pathway. We report a consanguineous family presenting with a hereditary motor neuropathy associated with a homozygous c.1A > G p.? variant of <i>COQ7</i> with abnormal CoQ<sub>10</sub> biosynthesis.</p><p><strong>Methods: </strong>Affected family members underwent clinical assessments that included nerve conduction testing, histologic analysis, and MRI. Pathogenicity of the <i>COQ7</i> variant was assessed in cultured fibroblasts and skeletal muscle using a combination of immunoblots, respirometry, and quinone analysis.</p><p><strong>Results: </strong>Three affected siblings, ranging from 12 to 24 years of age, presented with a severe length-dependent motor neuropathy with marked symmetric distal weakness and atrophy with normal sensation. Muscle biopsy of the quadriceps revealed chronic denervation pattern. An MRI examination identified moderate to severe fat infiltration in distal muscles. Exome sequencing demonstrated the homozygous <i>COQ7</i> c.1A > G p.? variant that is expected to bypass the first 38 amino acid residues at the n-terminus, initiating instead with methionine at position 39. This is predicted to cause the loss of the cleavable mitochondrial targeting sequence and 2 additional amino acids, thereby preventing the incorporation and subsequent folding of COQ7 into the inner mitochondrial membrane. Pathogenicity of the <i>COQ7</i> variant was demonstrated by diminished COQ7 and CoQ<sub>10</sub> levels in muscle and fibroblast samples of affected siblings but not in the father, unaffected sibling, or unrelated controls. In addition, fibroblasts from affected siblings had substantial accumulation of DMQ<sub>10</sub>, and maximal mitochondrial respiration was impaired in both fibroblasts and muscle.</p><p><strong>Discussion: </strong>This report describes a new neurologic phenotype of <i>COQ7</i>-related primary CoQ<sub>10</sub> deficiency. Novel aspects of the phenotype presented by this family include pure distal motor neuropathy involvement, as well as the lack of upper motor neuron features, cognitive delay, or sensory involvement in comparison with cases of <i>COQ7</i>-related CoQ<sub>10</sub> deficiency previously reported in the literature.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 1","pages":"e200048"},"PeriodicalIF":3.0000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/65/19/NXG-2022-200051.PMC10108386.pdf","citationCount":"2","resultStr":"{\"title\":\"Novel Homozygous Variant in <i>COQ7</i> in Siblings With Hereditary Motor Neuropathy.\",\"authors\":\"Ian C Smith, Chantal A Pileggi, Ying Wang, Kristin Kernohan, Taila Hartley, Hugh J McMillan, Marcos Loreto Sampaio, Gerd Melkus, John Woulfe, Gaganvir Parmar, Pierre R Bourque, Ari Breiner, Jocelyn Zwicker, C Elizabeth Pringle, Olga Jarinova, Hanns Lochmüller, David A Dyment, Bernard Brais, Kym M Boycott, Siegfried Hekimi, Mary-Ellen Harper, Jodi Warman-Chardon\",\"doi\":\"10.1212/NXG.0000000000200048\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong>Coenzyme Q<sub>10</sub> (CoQ<sub>10</sub>) is an important electron carrier and antioxidant. The COQ7 enzyme catalyzes the hydroxylation of 5-demethoxyubiquinone-10 (DMQ<sub>10</sub>), the second-to-last step in the CoQ<sub>10</sub> biosynthesis pathway. We report a consanguineous family presenting with a hereditary motor neuropathy associated with a homozygous c.1A > G p.? variant of <i>COQ7</i> with abnormal CoQ<sub>10</sub> biosynthesis.</p><p><strong>Methods: </strong>Affected family members underwent clinical assessments that included nerve conduction testing, histologic analysis, and MRI. Pathogenicity of the <i>COQ7</i> variant was assessed in cultured fibroblasts and skeletal muscle using a combination of immunoblots, respirometry, and quinone analysis.</p><p><strong>Results: </strong>Three affected siblings, ranging from 12 to 24 years of age, presented with a severe length-dependent motor neuropathy with marked symmetric distal weakness and atrophy with normal sensation. Muscle biopsy of the quadriceps revealed chronic denervation pattern. An MRI examination identified moderate to severe fat infiltration in distal muscles. Exome sequencing demonstrated the homozygous <i>COQ7</i> c.1A > G p.? variant that is expected to bypass the first 38 amino acid residues at the n-terminus, initiating instead with methionine at position 39. This is predicted to cause the loss of the cleavable mitochondrial targeting sequence and 2 additional amino acids, thereby preventing the incorporation and subsequent folding of COQ7 into the inner mitochondrial membrane. Pathogenicity of the <i>COQ7</i> variant was demonstrated by diminished COQ7 and CoQ<sub>10</sub> levels in muscle and fibroblast samples of affected siblings but not in the father, unaffected sibling, or unrelated controls. In addition, fibroblasts from affected siblings had substantial accumulation of DMQ<sub>10</sub>, and maximal mitochondrial respiration was impaired in both fibroblasts and muscle.</p><p><strong>Discussion: </strong>This report describes a new neurologic phenotype of <i>COQ7</i>-related primary CoQ<sub>10</sub> deficiency. Novel aspects of the phenotype presented by this family include pure distal motor neuropathy involvement, as well as the lack of upper motor neuron features, cognitive delay, or sensory involvement in comparison with cases of <i>COQ7</i>-related CoQ<sub>10</sub> deficiency previously reported in the literature.</p>\",\"PeriodicalId\":48613,\"journal\":{\"name\":\"Neurology-Genetics\",\"volume\":\"9 1\",\"pages\":\"e200048\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2023-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/65/19/NXG-2022-200051.PMC10108386.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology-Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1212/NXG.0000000000200048\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200048","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 2
摘要
背景与目的:辅酶Q10 (CoQ10)是一种重要的电子载体和抗氧化剂。辅酶q7酶催化5-去甲氧基双醌-10 (DMQ10)的羟基化,这是辅酶q10生物合成途径的倒数第二步。我们报告一个近亲家庭表现为遗传性运动神经病与纯合子c.1A > G . p.相关。辅酶q7的变异,辅酶q10的生物合成异常。方法:受影响的家庭成员接受临床评估,包括神经传导测试、组织学分析和MRI。COQ7变异的致病性在培养成纤维细胞和骨骼肌中进行评估,采用免疫印迹、呼吸测量和醌分析相结合的方法。结果:三名受影响的兄弟姐妹,年龄从12岁到24岁,表现为严重的长度依赖性运动神经病变,伴有明显的对称远端无力和萎缩,感觉正常。股四头肌的肌肉活检显示慢性去神经模式。MRI检查发现远端肌肉中中度至重度脂肪浸润。外显子组测序显示COQ7 c.1A > G . p.?该变异预计会绕过n端前38个氨基酸残基,以39号位置的蛋氨酸起始。预计这将导致可切割线粒体靶向序列和2个额外氨基酸的丢失,从而阻止COQ7进入线粒体内膜并随后折叠。COQ7变异的致病性是通过在患病兄弟姐妹的肌肉和成纤维细胞样本中降低COQ7和CoQ10水平来证明的,但在父亲、未患病兄弟姐妹或无关对照中没有。此外,来自受影响的兄弟姐妹的成纤维细胞有大量的DMQ10积累,成纤维细胞和肌肉的最大线粒体呼吸都受损。讨论:本报告描述了辅酶q7相关的原发性辅酶q10缺乏症的一种新的神经表型。该家族提出的表型的新方面包括纯远端运动神经病变受累,以及与文献中先前报道的coq7相关的CoQ10缺乏症相比,缺乏上运动神经元特征、认知延迟或感觉受累。
Novel Homozygous Variant in COQ7 in Siblings With Hereditary Motor Neuropathy.
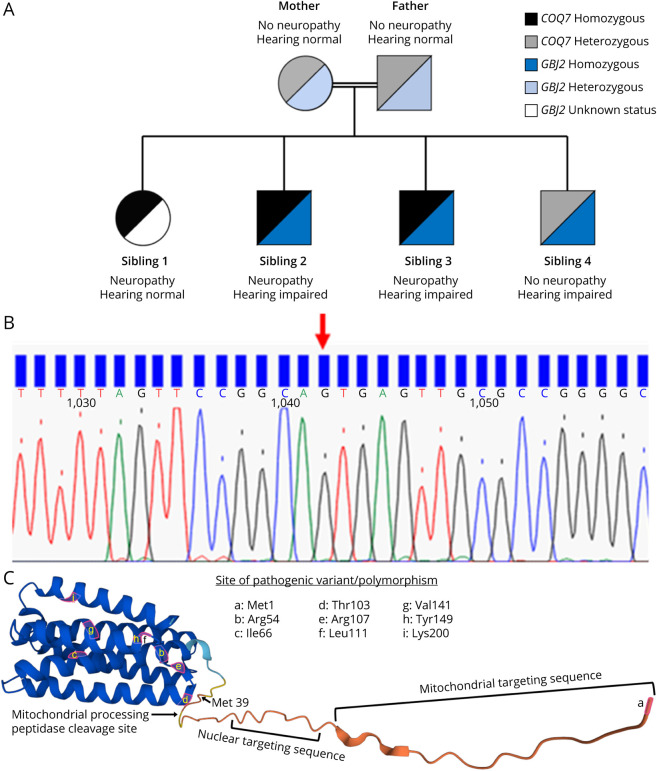
Background and objectives: Coenzyme Q10 (CoQ10) is an important electron carrier and antioxidant. The COQ7 enzyme catalyzes the hydroxylation of 5-demethoxyubiquinone-10 (DMQ10), the second-to-last step in the CoQ10 biosynthesis pathway. We report a consanguineous family presenting with a hereditary motor neuropathy associated with a homozygous c.1A > G p.? variant of COQ7 with abnormal CoQ10 biosynthesis.
Methods: Affected family members underwent clinical assessments that included nerve conduction testing, histologic analysis, and MRI. Pathogenicity of the COQ7 variant was assessed in cultured fibroblasts and skeletal muscle using a combination of immunoblots, respirometry, and quinone analysis.
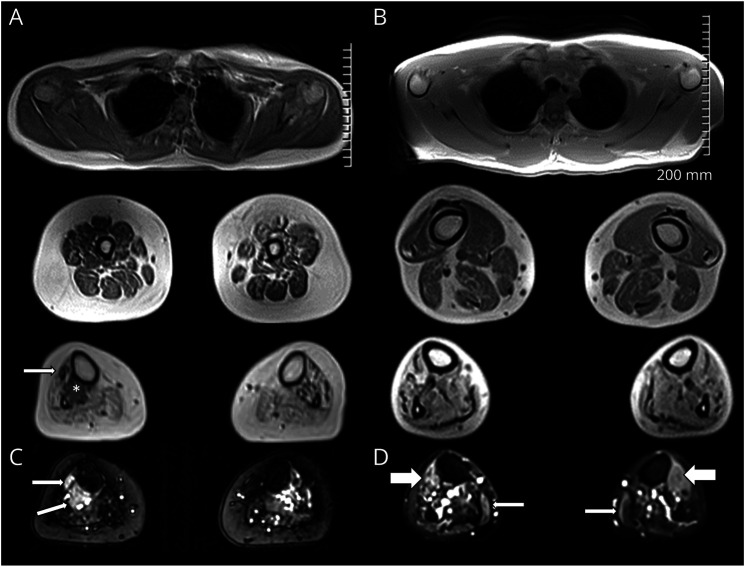
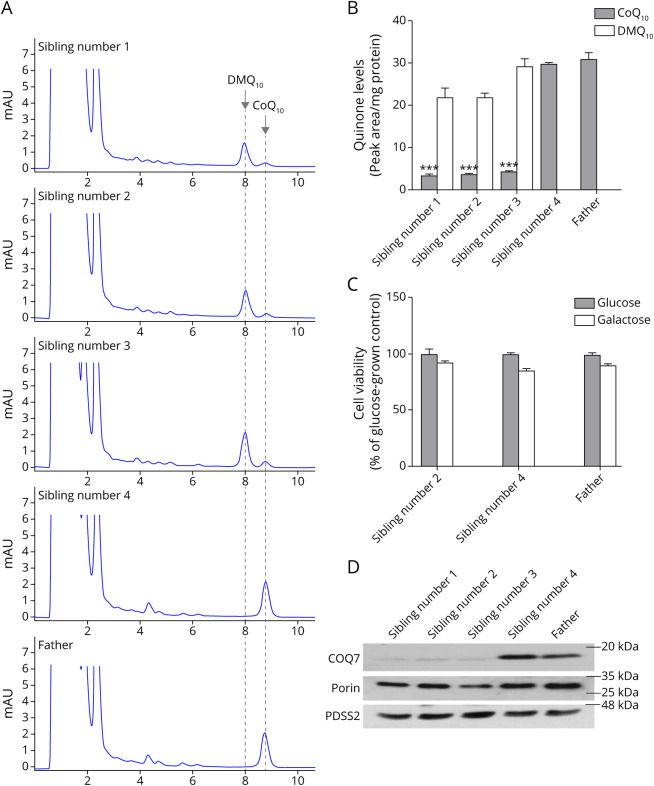
Results: Three affected siblings, ranging from 12 to 24 years of age, presented with a severe length-dependent motor neuropathy with marked symmetric distal weakness and atrophy with normal sensation. Muscle biopsy of the quadriceps revealed chronic denervation pattern. An MRI examination identified moderate to severe fat infiltration in distal muscles. Exome sequencing demonstrated the homozygous COQ7 c.1A > G p.? variant that is expected to bypass the first 38 amino acid residues at the n-terminus, initiating instead with methionine at position 39. This is predicted to cause the loss of the cleavable mitochondrial targeting sequence and 2 additional amino acids, thereby preventing the incorporation and subsequent folding of COQ7 into the inner mitochondrial membrane. Pathogenicity of the COQ7 variant was demonstrated by diminished COQ7 and CoQ10 levels in muscle and fibroblast samples of affected siblings but not in the father, unaffected sibling, or unrelated controls. In addition, fibroblasts from affected siblings had substantial accumulation of DMQ10, and maximal mitochondrial respiration was impaired in both fibroblasts and muscle.
Discussion: This report describes a new neurologic phenotype of COQ7-related primary CoQ10 deficiency. Novel aspects of the phenotype presented by this family include pure distal motor neuropathy involvement, as well as the lack of upper motor neuron features, cognitive delay, or sensory involvement in comparison with cases of COQ7-related CoQ10 deficiency previously reported in the literature.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


