Amanda P Smith, Lindey C Lane, Ivan Ramirez Zuniga, David M Moquin, Peter Vogel, Amber M Smith
{"title":"在流感相关的继发性细菌感染过程中,病毒传播的增加会导致肺损伤加重,但不会导致炎症。","authors":"Amanda P Smith, Lindey C Lane, Ivan Ramirez Zuniga, David M Moquin, Peter Vogel, Amber M Smith","doi":"10.1093/femsmc/xtac022","DOIUrl":null,"url":null,"abstract":"<p><p>Secondary bacterial infections increase influenza-related morbidity and mortality, particularly if acquired after 5-7 d from the viral onset. Synergistic host responses and direct pathogen-pathogen interactions are thought to lead to a state of hyperinflammation, but the kinetics of the lung pathology have not yet been detailed, and identifying the contribution of different mechanisms to disease is difficult because these may change over time. To address this gap, we examined host-pathogen and lung pathology dynamics following a secondary bacterial infection initiated at different time points after influenza within a murine model. We then used a mathematical approach to quantify the increased virus dissemination in the lung, coinfection time-dependent bacterial kinetics, and virus-mediated and postbacterial depletion of alveolar macrophages. The data showed that viral loads increase regardless of coinfection timing, which our mathematical model predicted and histomorphometry data confirmed was due to a robust increase in the number of infected cells. Bacterial loads were dependent on the time of coinfection and corresponded to the level of IAV-induced alveolar macrophage depletion. Our mathematical model suggested that the additional depletion of these cells following the bacterial invasion was mediated primarily by the virus. Contrary to current belief, inflammation was not enhanced and did not correlate with neutrophilia. The enhanced disease severity was correlated to inflammation, but this was due to a nonlinearity in this correlation. This study highlights the importance of dissecting nonlinearities during complex infections and demonstrated the increased dissemination of virus within the lung during bacterial coinfection and simultaneous modulation of immune responses during influenza-associated bacterial pneumonia.</p>","PeriodicalId":73024,"journal":{"name":"FEMS microbes","volume":"3 ","pages":"xtac022"},"PeriodicalIF":0.0000,"publicationDate":"2022-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/32/6e/xtac022.PMC10117793.pdf","citationCount":"0","resultStr":"{\"title\":\"Increased virus dissemination leads to enhanced lung injury but not inflammation during influenza-associated secondary bacterial infection.\",\"authors\":\"Amanda P Smith, Lindey C Lane, Ivan Ramirez Zuniga, David M Moquin, Peter Vogel, Amber M Smith\",\"doi\":\"10.1093/femsmc/xtac022\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Secondary bacterial infections increase influenza-related morbidity and mortality, particularly if acquired after 5-7 d from the viral onset. Synergistic host responses and direct pathogen-pathogen interactions are thought to lead to a state of hyperinflammation, but the kinetics of the lung pathology have not yet been detailed, and identifying the contribution of different mechanisms to disease is difficult because these may change over time. To address this gap, we examined host-pathogen and lung pathology dynamics following a secondary bacterial infection initiated at different time points after influenza within a murine model. We then used a mathematical approach to quantify the increased virus dissemination in the lung, coinfection time-dependent bacterial kinetics, and virus-mediated and postbacterial depletion of alveolar macrophages. The data showed that viral loads increase regardless of coinfection timing, which our mathematical model predicted and histomorphometry data confirmed was due to a robust increase in the number of infected cells. Bacterial loads were dependent on the time of coinfection and corresponded to the level of IAV-induced alveolar macrophage depletion. Our mathematical model suggested that the additional depletion of these cells following the bacterial invasion was mediated primarily by the virus. Contrary to current belief, inflammation was not enhanced and did not correlate with neutrophilia. The enhanced disease severity was correlated to inflammation, but this was due to a nonlinearity in this correlation. This study highlights the importance of dissecting nonlinearities during complex infections and demonstrated the increased dissemination of virus within the lung during bacterial coinfection and simultaneous modulation of immune responses during influenza-associated bacterial pneumonia.</p>\",\"PeriodicalId\":73024,\"journal\":{\"name\":\"FEMS microbes\",\"volume\":\"3 \",\"pages\":\"xtac022\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-07-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/32/6e/xtac022.PMC10117793.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"FEMS microbes\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/femsmc/xtac022\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"FEMS microbes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/femsmc/xtac022","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Increased virus dissemination leads to enhanced lung injury but not inflammation during influenza-associated secondary bacterial infection.
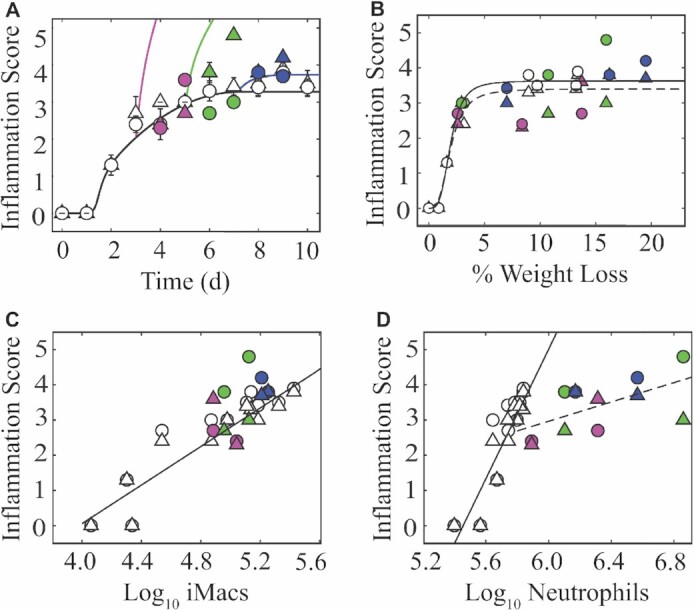
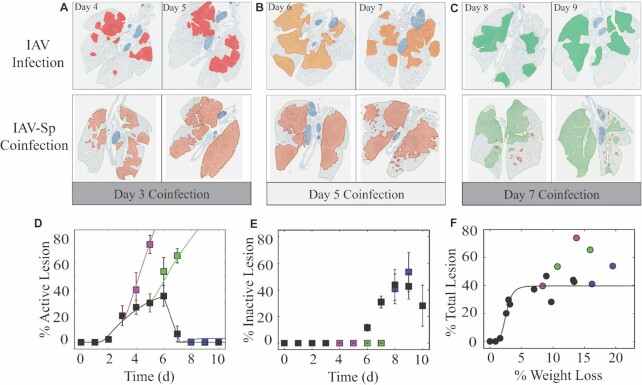
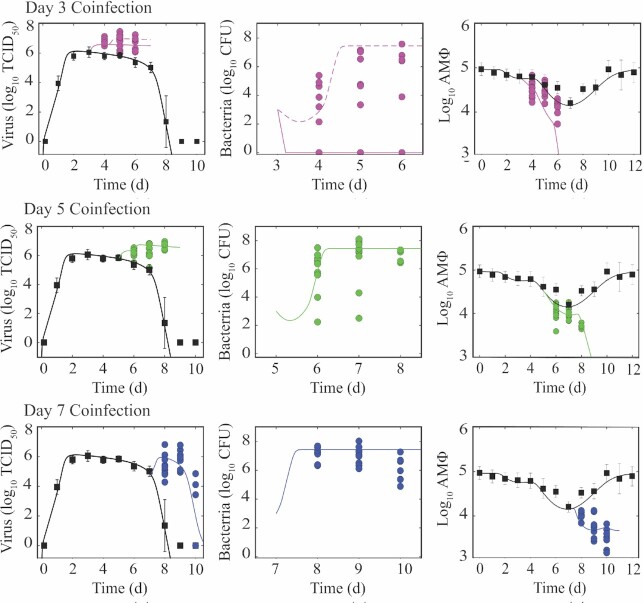
Secondary bacterial infections increase influenza-related morbidity and mortality, particularly if acquired after 5-7 d from the viral onset. Synergistic host responses and direct pathogen-pathogen interactions are thought to lead to a state of hyperinflammation, but the kinetics of the lung pathology have not yet been detailed, and identifying the contribution of different mechanisms to disease is difficult because these may change over time. To address this gap, we examined host-pathogen and lung pathology dynamics following a secondary bacterial infection initiated at different time points after influenza within a murine model. We then used a mathematical approach to quantify the increased virus dissemination in the lung, coinfection time-dependent bacterial kinetics, and virus-mediated and postbacterial depletion of alveolar macrophages. The data showed that viral loads increase regardless of coinfection timing, which our mathematical model predicted and histomorphometry data confirmed was due to a robust increase in the number of infected cells. Bacterial loads were dependent on the time of coinfection and corresponded to the level of IAV-induced alveolar macrophage depletion. Our mathematical model suggested that the additional depletion of these cells following the bacterial invasion was mediated primarily by the virus. Contrary to current belief, inflammation was not enhanced and did not correlate with neutrophilia. The enhanced disease severity was correlated to inflammation, but this was due to a nonlinearity in this correlation. This study highlights the importance of dissecting nonlinearities during complex infections and demonstrated the increased dissemination of virus within the lung during bacterial coinfection and simultaneous modulation of immune responses during influenza-associated bacterial pneumonia.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


