{"title":"如何在深度蛋白质组学数据分析中使用open-pFind ?-从质谱数据中严格鉴定和定量肽和蛋白质的协议。","authors":"Guangcan Shao, Yong Cao, Zhenlin Chen, Chao Liu, Shangtong Li, Hao Chi, Meng-Qiu Dong","doi":"10.52601/bpr.2021.210004","DOIUrl":null,"url":null,"abstract":"<p><p>High-throughput proteomics based on mass spectrometry (MS) analysis has permeated biomedical science and propelled numerous research projects. pFind 3 is a database search engine for high-speed and in-depth proteomics data analysis. pFind 3 features a swift open search workflow that is adept at uncovering less obvious information such as unexpected modifications or mutations that would have gone unnoticed using a conventional data analysis pipeline. In this protocol, we provide step-by-step instructions to help users mastering various types of data analysis using pFind 3 in conjunction with pParse for data pre-processing and if needed, pQuant for quantitation. This streamlined pParse-pFind-pQuant workflow offers exceptional sensitivity, precision, and speed. It can be easily implemented in any laboratory in need of identifying peptides, proteins, or post-translational modifications, or of quantitation based on <sup>15</sup>N-labeling, SILAC-labeling, or TMT/iTRAQ labeling.</p>","PeriodicalId":59621,"journal":{"name":"生物物理学报:英文版","volume":"7 3","pages":"207-226"},"PeriodicalIF":0.0000,"publicationDate":"2021-06-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10244800/pdf/","citationCount":"10","resultStr":"{\"title\":\"How to use open-pFind in deep proteomics data analysis?- A protocol for rigorous identification and quantitation of peptides and proteins from mass spectrometry data.\",\"authors\":\"Guangcan Shao, Yong Cao, Zhenlin Chen, Chao Liu, Shangtong Li, Hao Chi, Meng-Qiu Dong\",\"doi\":\"10.52601/bpr.2021.210004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>High-throughput proteomics based on mass spectrometry (MS) analysis has permeated biomedical science and propelled numerous research projects. pFind 3 is a database search engine for high-speed and in-depth proteomics data analysis. pFind 3 features a swift open search workflow that is adept at uncovering less obvious information such as unexpected modifications or mutations that would have gone unnoticed using a conventional data analysis pipeline. In this protocol, we provide step-by-step instructions to help users mastering various types of data analysis using pFind 3 in conjunction with pParse for data pre-processing and if needed, pQuant for quantitation. This streamlined pParse-pFind-pQuant workflow offers exceptional sensitivity, precision, and speed. It can be easily implemented in any laboratory in need of identifying peptides, proteins, or post-translational modifications, or of quantitation based on <sup>15</sup>N-labeling, SILAC-labeling, or TMT/iTRAQ labeling.</p>\",\"PeriodicalId\":59621,\"journal\":{\"name\":\"生物物理学报:英文版\",\"volume\":\"7 3\",\"pages\":\"207-226\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-06-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10244800/pdf/\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"生物物理学报:英文版\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.52601/bpr.2021.210004\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"生物物理学报:英文版","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.52601/bpr.2021.210004","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
How to use open-pFind in deep proteomics data analysis?- A protocol for rigorous identification and quantitation of peptides and proteins from mass spectrometry data.
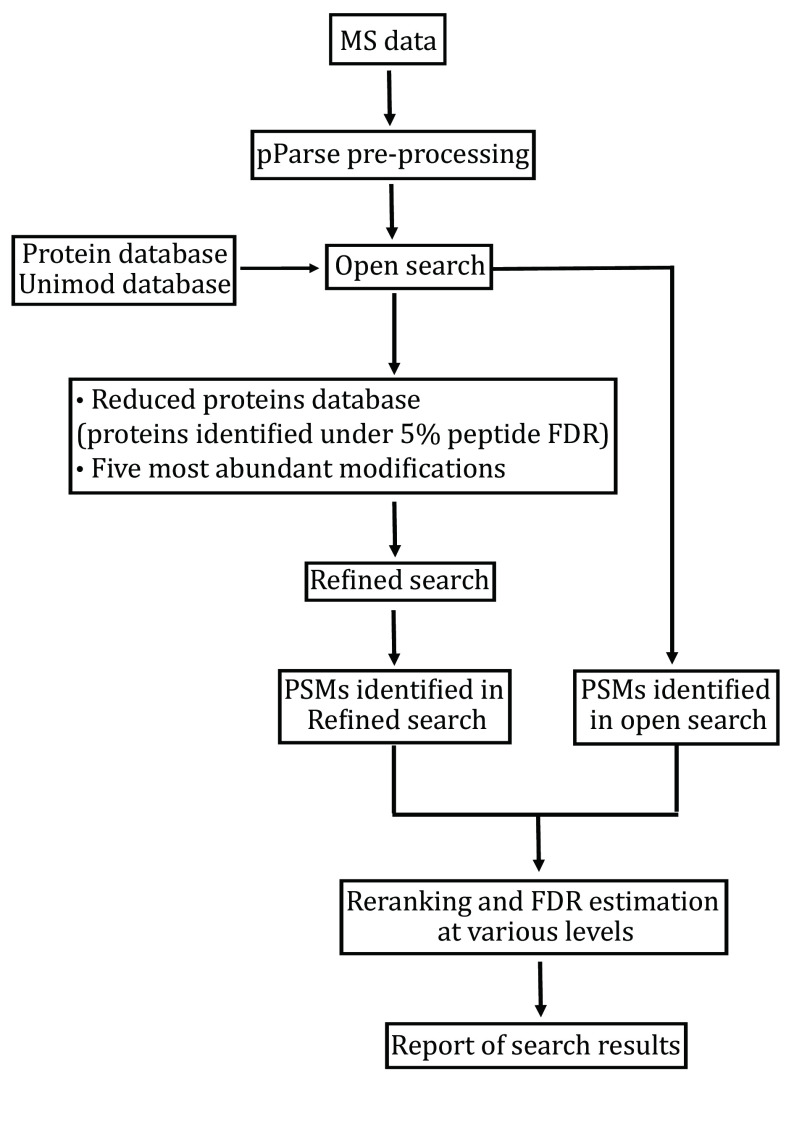
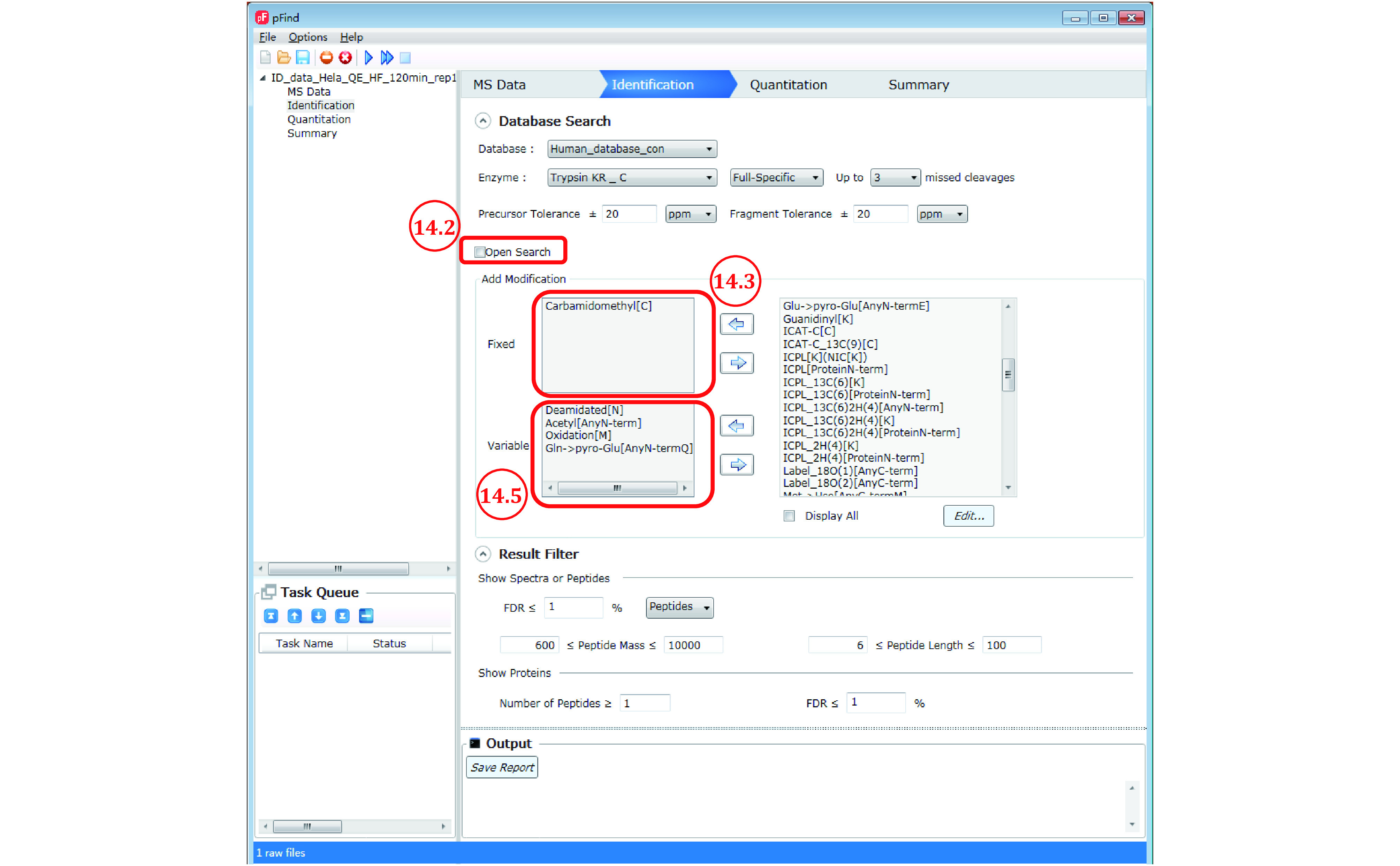
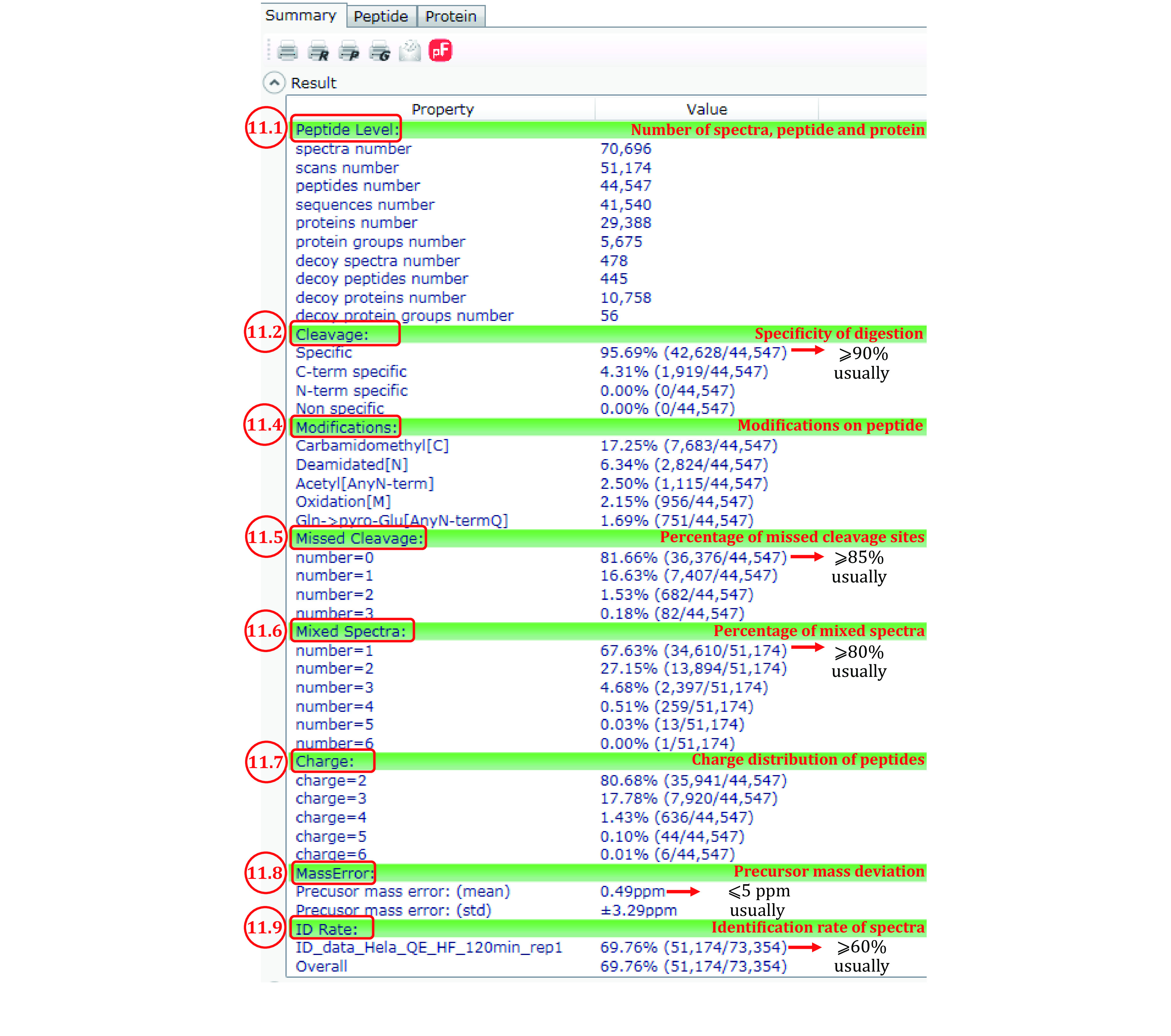
High-throughput proteomics based on mass spectrometry (MS) analysis has permeated biomedical science and propelled numerous research projects. pFind 3 is a database search engine for high-speed and in-depth proteomics data analysis. pFind 3 features a swift open search workflow that is adept at uncovering less obvious information such as unexpected modifications or mutations that would have gone unnoticed using a conventional data analysis pipeline. In this protocol, we provide step-by-step instructions to help users mastering various types of data analysis using pFind 3 in conjunction with pParse for data pre-processing and if needed, pQuant for quantitation. This streamlined pParse-pFind-pQuant workflow offers exceptional sensitivity, precision, and speed. It can be easily implemented in any laboratory in need of identifying peptides, proteins, or post-translational modifications, or of quantitation based on 15N-labeling, SILAC-labeling, or TMT/iTRAQ labeling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


