{"title":"两家系III型糖原储存病患者新型AGL变异的临床和功能特征","authors":"Tingting Yu, Hao Fu, Aoyu Yang, Yan Liang","doi":"10.1155/2023/6679871","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Glycogen storage disease type III (GSDIII) is a uncommon autosomal recessive inherited metabolic disorder, which is caused by variants in the AGL gene. The purpose of this study was to elucidate the clinical and functional features of two novel variants in two families with GSDIIIa.</p><p><strong>Methods: </strong>We collected the clinical and laboratory data of the two patients. Genetic testing was performed using GSDs gene panel sequencing, and the identified variants were classified according to the American College of Medical Genetics (ACMG) criteria. The pathogenicity of the novel variants was furthermore assessed through bioinformatics analysis and cellular functional validation experiments.</p><p><strong>Results: </strong>The two patients were hospitalized with abnormal liver function or hepatomegaly, which was characterized by remarkably elevated liver enzyme and muscle enzyme levels, as well as hepatomegaly, and were eventually diagnosed with GSDIIIa. Genetic analysis detected two novel variants of AGL gene in the two patients: c.1484A > G (p.Y495C), c.1981G > T (p.D661Y). Bioinformatics analysis indicated that the two novel missense mutations most likely altered the protein's conformation and therefore made the enzyme it encodes less active. Based on the ACMG criteria, both variants were considered likely pathogenic, in accordance with the functional analysis results, which demonstrated that the mutated protein was still localized in the cytoplasm and that the glycogen content of cells transfected with the mutated AGL was increased compared to cells transfected with the wild-type one.</p><p><strong>Conclusion: </strong>These findings indicated that the two newly identified variants in the AGL gene (c.1484A > G; c.1981G > T) were undoubtedly pathogenic mutations, inducing a slight reduction in glycogen debranching enzyme activity and a mild increase in intracellular glycogen content. Two patients who visited us with abnormal liver function, or hepatomegaly, improved dramatically after treatment with oral uncooked cornstarch, but the effects on skeletal muscle and myocardium required further observation.</p>","PeriodicalId":13966,"journal":{"name":"International Journal of Endocrinology","volume":"2023 ","pages":"6679871"},"PeriodicalIF":2.3000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10243941/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical and Functional Characterization of Novel AGL Variants in Two Families with Glycogen Storage Disease Type III.\",\"authors\":\"Tingting Yu, Hao Fu, Aoyu Yang, Yan Liang\",\"doi\":\"10.1155/2023/6679871\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose: </strong>Glycogen storage disease type III (GSDIII) is a uncommon autosomal recessive inherited metabolic disorder, which is caused by variants in the AGL gene. The purpose of this study was to elucidate the clinical and functional features of two novel variants in two families with GSDIIIa.</p><p><strong>Methods: </strong>We collected the clinical and laboratory data of the two patients. Genetic testing was performed using GSDs gene panel sequencing, and the identified variants were classified according to the American College of Medical Genetics (ACMG) criteria. The pathogenicity of the novel variants was furthermore assessed through bioinformatics analysis and cellular functional validation experiments.</p><p><strong>Results: </strong>The two patients were hospitalized with abnormal liver function or hepatomegaly, which was characterized by remarkably elevated liver enzyme and muscle enzyme levels, as well as hepatomegaly, and were eventually diagnosed with GSDIIIa. Genetic analysis detected two novel variants of AGL gene in the two patients: c.1484A > G (p.Y495C), c.1981G > T (p.D661Y). Bioinformatics analysis indicated that the two novel missense mutations most likely altered the protein's conformation and therefore made the enzyme it encodes less active. Based on the ACMG criteria, both variants were considered likely pathogenic, in accordance with the functional analysis results, which demonstrated that the mutated protein was still localized in the cytoplasm and that the glycogen content of cells transfected with the mutated AGL was increased compared to cells transfected with the wild-type one.</p><p><strong>Conclusion: </strong>These findings indicated that the two newly identified variants in the AGL gene (c.1484A > G; c.1981G > T) were undoubtedly pathogenic mutations, inducing a slight reduction in glycogen debranching enzyme activity and a mild increase in intracellular glycogen content. Two patients who visited us with abnormal liver function, or hepatomegaly, improved dramatically after treatment with oral uncooked cornstarch, but the effects on skeletal muscle and myocardium required further observation.</p>\",\"PeriodicalId\":13966,\"journal\":{\"name\":\"International Journal of Endocrinology\",\"volume\":\"2023 \",\"pages\":\"6679871\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10243941/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Endocrinology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/6679871\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Endocrinology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1155/2023/6679871","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
目的:糖原储存病III型(GSDIII)是一种罕见的常染色体隐性遗传代谢疾病,由AGL基因变异引起。本研究的目的是阐明两个GSDIIIa家族中两个新变异的临床和功能特征。方法:收集2例患者的临床及实验室资料。采用GSDs基因面板测序进行基因检测,并根据美国医学遗传学学院(American College of Medical Genetics, ACMG)标准对鉴定的变异进行分类。通过生物信息学分析和细胞功能验证实验进一步评估了新变异的致病性。结果:2例患者住院时均出现肝功能异常或肝肿大,表现为肝酶、肌酶水平明显升高,并伴有肝肿大,最终诊断为GSDIIIa。遗传分析在2例患者中检测到两个新的AGL基因变异:c.1484A > G (p.Y495C), c.1981G > T (p.D661Y)。生物信息学分析表明,这两个新的错义突变很可能改变了蛋白质的构象,从而使其编码的酶活性降低。根据ACMG标准,根据功能分析结果,这两个变异都被认为是可能致病的,这表明突变蛋白仍然定位在细胞质中,并且与转染野生型相比,转染突变AGL的细胞的糖原含量增加。结论:新发现的2个AGL基因变异(c.1484A > G;c.1981G > T)无疑是致病性突变,引起糖原脱分枝酶活性的轻微降低和细胞内糖原含量的轻微增加。2例肝功能异常或肝肿大患者经口服生玉米淀粉治疗后明显改善,但对骨骼肌和心肌的影响有待进一步观察。
Clinical and Functional Characterization of Novel AGL Variants in Two Families with Glycogen Storage Disease Type III.
Purpose: Glycogen storage disease type III (GSDIII) is a uncommon autosomal recessive inherited metabolic disorder, which is caused by variants in the AGL gene. The purpose of this study was to elucidate the clinical and functional features of two novel variants in two families with GSDIIIa.
Methods: We collected the clinical and laboratory data of the two patients. Genetic testing was performed using GSDs gene panel sequencing, and the identified variants were classified according to the American College of Medical Genetics (ACMG) criteria. The pathogenicity of the novel variants was furthermore assessed through bioinformatics analysis and cellular functional validation experiments.
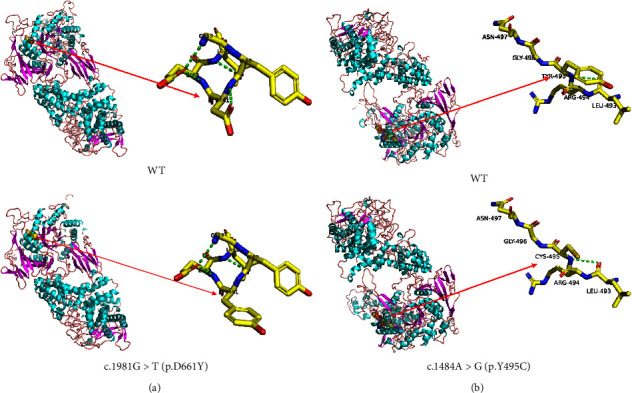
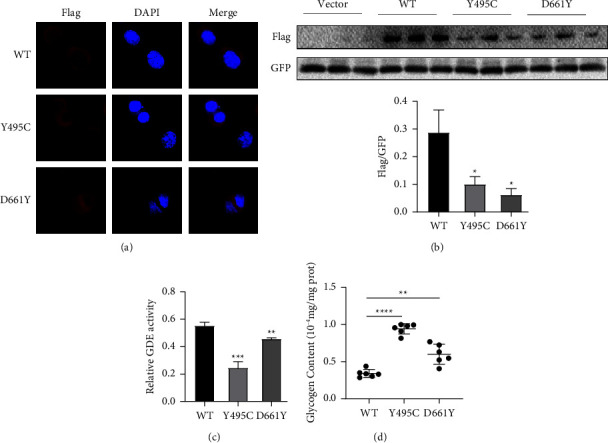
Results: The two patients were hospitalized with abnormal liver function or hepatomegaly, which was characterized by remarkably elevated liver enzyme and muscle enzyme levels, as well as hepatomegaly, and were eventually diagnosed with GSDIIIa. Genetic analysis detected two novel variants of AGL gene in the two patients: c.1484A > G (p.Y495C), c.1981G > T (p.D661Y). Bioinformatics analysis indicated that the two novel missense mutations most likely altered the protein's conformation and therefore made the enzyme it encodes less active. Based on the ACMG criteria, both variants were considered likely pathogenic, in accordance with the functional analysis results, which demonstrated that the mutated protein was still localized in the cytoplasm and that the glycogen content of cells transfected with the mutated AGL was increased compared to cells transfected with the wild-type one.
Conclusion: These findings indicated that the two newly identified variants in the AGL gene (c.1484A > G; c.1981G > T) were undoubtedly pathogenic mutations, inducing a slight reduction in glycogen debranching enzyme activity and a mild increase in intracellular glycogen content. Two patients who visited us with abnormal liver function, or hepatomegaly, improved dramatically after treatment with oral uncooked cornstarch, but the effects on skeletal muscle and myocardium required further observation.
期刊介绍:
International Journal of Endocrinology is a peer-reviewed, Open Access journal that provides a forum for scientists and clinicians working in basic and translational research. The journal publishes original research articles, review articles, and clinical studies that provide insights into the endocrine system and its associated diseases at a genomic, molecular, biochemical and cellular level.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


