{"title":"台湾威尔森氏病的临床特征、遗传特征和长期预后:一项11年的随访研究。","authors":"Sung-Pin Fan, Yih-Chih Kuo, Ni-Chung Lee, Yin-Hsiu Chien, Wuh-Liang Hwu, Yu-Hsuan Huang, Han-I Lin, Tai-Chung Tseng, Tung-Hung Su, Shiou-Ru Tzeng, Chien-Ting Hsu, Huey-Ling Chen, Chin-Hsien Lin, Yen-Hsuan Ni","doi":"10.14802/jmd.22161","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>aaWilson's disease (WD) is a rare genetic disorder of copper metabolism, and longitudinal follow-up studies are limited. We performed a retrospective analysis to determine the clinical characteristics and long-term outcomes in a large WD cohort.</p><p><strong>Methods: </strong>aaMedical records of WD patients diagnosed from 2006-2021 at National Taiwan University Hospital were retrospectively evaluated for clinical presentations, neuroimages, genetic information, and follow-up outcomes.</p><p><strong>Results: </strong>aaThe present study enrolled 123 WD patients (mean follow-up: 11.12 ± 7.41 years), including 74 patients (60.2%) with hepatic features and 49 patients (39.8%) with predominantly neuropsychiatric symptoms. Compared to the hepatic group, the neuropsychiatric group exhibited more Kayser-Fleischer rings (77.6% vs. 41.9%, p < 0.01), lower serum ceruloplasmin levels (4.9 ± 3.9 vs. 6.3 ± 3.9 mg/dL, p < 0.01), smaller total brain and subcortical gray matter volumes (p < 0.0001), and worse functional outcomes during follow-up (p = 0.0003). Among patients with available DNA samples (n = 59), the most common mutations were p.R778L (allelic frequency of 22.03%) followed by p.P992L (11.86%) and p.T935M (9.32%). Patients with at least one allele of p.R778L had a younger onset age (p = 0.04), lower ceruloplasmin levels (p < 0.01), lower serum copper levels (p = 0.03), higher percentage of the hepatic form (p = 0.03), and a better functional outcome during follow-up (p = 0.0012) compared to patients with other genetic variations.</p><p><strong>Conclusion: </strong>aaThe distinct clinical characteristics and long-term outcomes of patients in our cohort support the ethnic differences regarding the mutational spectrum and clinical presentations in WD.</p>","PeriodicalId":2,"journal":{"name":"ACS Applied Bio Materials","volume":null,"pages":null},"PeriodicalIF":4.6000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ec/4e/jmd-22161.PMC10236021.pdf","citationCount":"1","resultStr":"{\"title\":\"Clinical Characteristics, Genetic Features, and Long-Term Outcome of Wilson's Disease in a Taiwanese Population: An 11-Year Follow-Up Study.\",\"authors\":\"Sung-Pin Fan, Yih-Chih Kuo, Ni-Chung Lee, Yin-Hsiu Chien, Wuh-Liang Hwu, Yu-Hsuan Huang, Han-I Lin, Tai-Chung Tseng, Tung-Hung Su, Shiou-Ru Tzeng, Chien-Ting Hsu, Huey-Ling Chen, Chin-Hsien Lin, Yen-Hsuan Ni\",\"doi\":\"10.14802/jmd.22161\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>aaWilson's disease (WD) is a rare genetic disorder of copper metabolism, and longitudinal follow-up studies are limited. We performed a retrospective analysis to determine the clinical characteristics and long-term outcomes in a large WD cohort.</p><p><strong>Methods: </strong>aaMedical records of WD patients diagnosed from 2006-2021 at National Taiwan University Hospital were retrospectively evaluated for clinical presentations, neuroimages, genetic information, and follow-up outcomes.</p><p><strong>Results: </strong>aaThe present study enrolled 123 WD patients (mean follow-up: 11.12 ± 7.41 years), including 74 patients (60.2%) with hepatic features and 49 patients (39.8%) with predominantly neuropsychiatric symptoms. Compared to the hepatic group, the neuropsychiatric group exhibited more Kayser-Fleischer rings (77.6% vs. 41.9%, p < 0.01), lower serum ceruloplasmin levels (4.9 ± 3.9 vs. 6.3 ± 3.9 mg/dL, p < 0.01), smaller total brain and subcortical gray matter volumes (p < 0.0001), and worse functional outcomes during follow-up (p = 0.0003). Among patients with available DNA samples (n = 59), the most common mutations were p.R778L (allelic frequency of 22.03%) followed by p.P992L (11.86%) and p.T935M (9.32%). Patients with at least one allele of p.R778L had a younger onset age (p = 0.04), lower ceruloplasmin levels (p < 0.01), lower serum copper levels (p = 0.03), higher percentage of the hepatic form (p = 0.03), and a better functional outcome during follow-up (p = 0.0012) compared to patients with other genetic variations.</p><p><strong>Conclusion: </strong>aaThe distinct clinical characteristics and long-term outcomes of patients in our cohort support the ethnic differences regarding the mutational spectrum and clinical presentations in WD.</p>\",\"PeriodicalId\":2,\"journal\":{\"name\":\"ACS Applied Bio Materials\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ec/4e/jmd-22161.PMC10236021.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Applied Bio Materials\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.14802/jmd.22161\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, BIOMATERIALS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Bio Materials","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.14802/jmd.22161","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, BIOMATERIALS","Score":null,"Total":0}
引用次数: 1
摘要
目的:威尔森病(WD)是一种罕见的铜代谢遗传性疾病,其纵向随访研究有限。我们进行了一项回顾性分析,以确定大型WD队列的临床特征和长期结果。方法:回顾性评价台大医院2006-2021年诊断的WD患者的临床表现、神经影像、遗传信息和随访结果。结果:本研究纳入123例WD患者(平均随访时间:11.12±7.41年),其中74例(60.2%)有肝脏特征,49例(39.8%)以神经精神症状为主。与肝脏组相比,神经精神组表现出更多的Kayser-Fleischer环(77.6% vs. 41.9%, p < 0.01),血清铜蓝蛋白水平较低(4.9±3.9 vs. 6.3±3.9 mg/dL, p < 0.01),脑和皮质下灰质总量较小(p < 0.0001),随访期间功能预后较差(p = 0.0003)。在可获得DNA样本的患者(n = 59)中,p.R778L最常见(等位基因频率为22.03%),其次是p.p 9992l(11.86%)和p.T935M(9.32%)。与其他遗传变异的患者相比,携带至少一个p.R778L等位基因的患者发病年龄更年轻(p = 0.04),铜蓝蛋白水平更低(p < 0.01),血清铜水平更低(p = 0.03),肝型比例更高(p = 0.03),随访期间功能预后更好(p = 0.0012)。结论:在我们的队列中,患者的不同临床特征和长期结局支持了在WD的突变谱和临床表现方面的种族差异。
Clinical Characteristics, Genetic Features, and Long-Term Outcome of Wilson's Disease in a Taiwanese Population: An 11-Year Follow-Up Study.
Objective: aaWilson's disease (WD) is a rare genetic disorder of copper metabolism, and longitudinal follow-up studies are limited. We performed a retrospective analysis to determine the clinical characteristics and long-term outcomes in a large WD cohort.
Methods: aaMedical records of WD patients diagnosed from 2006-2021 at National Taiwan University Hospital were retrospectively evaluated for clinical presentations, neuroimages, genetic information, and follow-up outcomes.
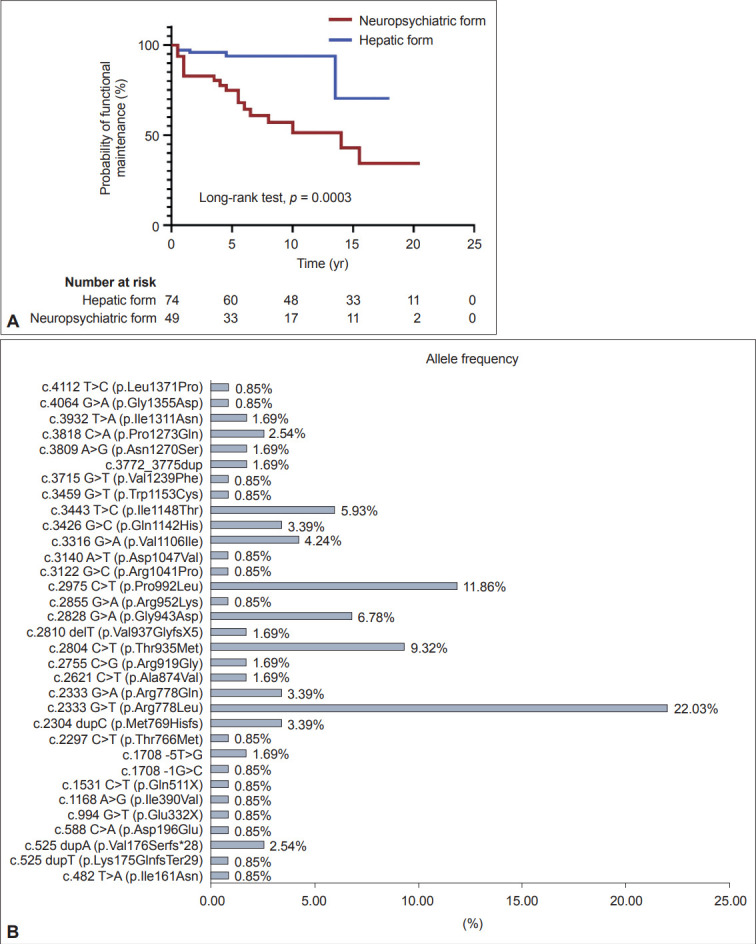
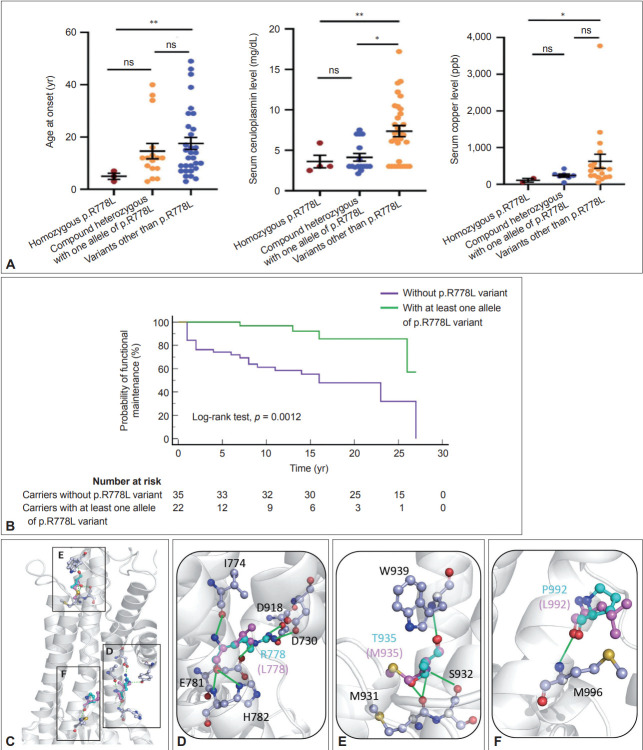
Results: aaThe present study enrolled 123 WD patients (mean follow-up: 11.12 ± 7.41 years), including 74 patients (60.2%) with hepatic features and 49 patients (39.8%) with predominantly neuropsychiatric symptoms. Compared to the hepatic group, the neuropsychiatric group exhibited more Kayser-Fleischer rings (77.6% vs. 41.9%, p < 0.01), lower serum ceruloplasmin levels (4.9 ± 3.9 vs. 6.3 ± 3.9 mg/dL, p < 0.01), smaller total brain and subcortical gray matter volumes (p < 0.0001), and worse functional outcomes during follow-up (p = 0.0003). Among patients with available DNA samples (n = 59), the most common mutations were p.R778L (allelic frequency of 22.03%) followed by p.P992L (11.86%) and p.T935M (9.32%). Patients with at least one allele of p.R778L had a younger onset age (p = 0.04), lower ceruloplasmin levels (p < 0.01), lower serum copper levels (p = 0.03), higher percentage of the hepatic form (p = 0.03), and a better functional outcome during follow-up (p = 0.0012) compared to patients with other genetic variations.
Conclusion: aaThe distinct clinical characteristics and long-term outcomes of patients in our cohort support the ethnic differences regarding the mutational spectrum and clinical presentations in WD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


