Leslie Hotchkiss Hayes, Morgane Perdomini, Asli Aykanat, Casie A Genetti, Heather L Paterson, Belinda S Cowling, Christian Freitag, Alan H Beggs
{"title":"dnm2相关核中性肌病的表型谱。","authors":"Leslie Hotchkiss Hayes, Morgane Perdomini, Asli Aykanat, Casie A Genetti, Heather L Paterson, Belinda S Cowling, Christian Freitag, Alan H Beggs","doi":"10.1212/NXG.0000000000200027","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Centronuclear myopathy (CNM) due to mutations in the dynamin 2 gene, <i>DNM2</i>, is a rare neuromuscular disease about which little is known. The objective of this study was to describe the range of clinical presentations and subsequent natural history of <i>DNM2</i>-related CNM.</p><p><strong>Methods: </strong>Pediatric and adult patients with suspicion for a CNM diagnosis and confirmed heterozygous pathogenic variants in <i>DNM2</i> were ascertained between December 8, 2000, and May 1, 2019. Data were collected through a retrospective review of genetic testing results, clinical records, and pathology slides combined with patient-reported clinical findings via questionnaires.</p><p><strong>Results: </strong>Forty-two patients with <i>DNM2</i>-related CNM, whose ages ranged from 0.95 to 75.76 years at most recent contact, were enrolled from 34 families in North or South America and Europe. There were 8 different <i>DNM2</i> pathogenic variants within the cohort. Of the 32 biopsied patients, all had histologic features of CNM. The disease onset was in infancy or childhood in 81% of the cohort, and more than half of the patients had high arched palates, indicative of weakness in utero. Ambulation was affected in nearly all (92%) the patients, and while the rapidity of progression was variable, most (67%) reported a \"deteriorating course.\" Ptosis, ophthalmoparesis, facial weakness, dysphagia, and respiratory insufficiency were commonly reported. One-third of the patients experienced restricted jaw mobility. Certain pathogenic variants appear to correlate with a more severe phenotype.</p><p><strong>Discussion: </strong><i>DNM2</i>-related CNM has a predominantly early-onset, often congenital, myopathy resulting in progressive difficulty with ambulation and occasionally bulbar and respiratory dysfunction. This detailed characterization of the phenotype provides important information to support clinical trial readiness for future disease-modifying therapies.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"8 6","pages":"e200027"},"PeriodicalIF":3.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/98/NXG-2022-200030.PMC9621335.pdf","citationCount":"2","resultStr":"{\"title\":\"Phenotypic Spectrum of <i>DNM2</i>-Related Centronuclear Myopathy.\",\"authors\":\"Leslie Hotchkiss Hayes, Morgane Perdomini, Asli Aykanat, Casie A Genetti, Heather L Paterson, Belinda S Cowling, Christian Freitag, Alan H Beggs\",\"doi\":\"10.1212/NXG.0000000000200027\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong>Centronuclear myopathy (CNM) due to mutations in the dynamin 2 gene, <i>DNM2</i>, is a rare neuromuscular disease about which little is known. The objective of this study was to describe the range of clinical presentations and subsequent natural history of <i>DNM2</i>-related CNM.</p><p><strong>Methods: </strong>Pediatric and adult patients with suspicion for a CNM diagnosis and confirmed heterozygous pathogenic variants in <i>DNM2</i> were ascertained between December 8, 2000, and May 1, 2019. Data were collected through a retrospective review of genetic testing results, clinical records, and pathology slides combined with patient-reported clinical findings via questionnaires.</p><p><strong>Results: </strong>Forty-two patients with <i>DNM2</i>-related CNM, whose ages ranged from 0.95 to 75.76 years at most recent contact, were enrolled from 34 families in North or South America and Europe. There were 8 different <i>DNM2</i> pathogenic variants within the cohort. Of the 32 biopsied patients, all had histologic features of CNM. The disease onset was in infancy or childhood in 81% of the cohort, and more than half of the patients had high arched palates, indicative of weakness in utero. Ambulation was affected in nearly all (92%) the patients, and while the rapidity of progression was variable, most (67%) reported a \\\"deteriorating course.\\\" Ptosis, ophthalmoparesis, facial weakness, dysphagia, and respiratory insufficiency were commonly reported. One-third of the patients experienced restricted jaw mobility. Certain pathogenic variants appear to correlate with a more severe phenotype.</p><p><strong>Discussion: </strong><i>DNM2</i>-related CNM has a predominantly early-onset, often congenital, myopathy resulting in progressive difficulty with ambulation and occasionally bulbar and respiratory dysfunction. This detailed characterization of the phenotype provides important information to support clinical trial readiness for future disease-modifying therapies.</p>\",\"PeriodicalId\":48613,\"journal\":{\"name\":\"Neurology-Genetics\",\"volume\":\"8 6\",\"pages\":\"e200027\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/98/NXG-2022-200030.PMC9621335.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology-Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1212/NXG.0000000000200027\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200027","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Phenotypic Spectrum of DNM2-Related Centronuclear Myopathy.
Background and objectives: Centronuclear myopathy (CNM) due to mutations in the dynamin 2 gene, DNM2, is a rare neuromuscular disease about which little is known. The objective of this study was to describe the range of clinical presentations and subsequent natural history of DNM2-related CNM.
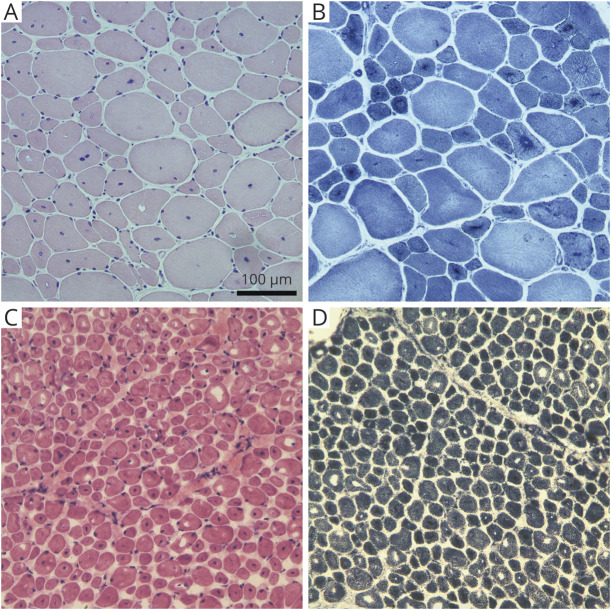
Methods: Pediatric and adult patients with suspicion for a CNM diagnosis and confirmed heterozygous pathogenic variants in DNM2 were ascertained between December 8, 2000, and May 1, 2019. Data were collected through a retrospective review of genetic testing results, clinical records, and pathology slides combined with patient-reported clinical findings via questionnaires.
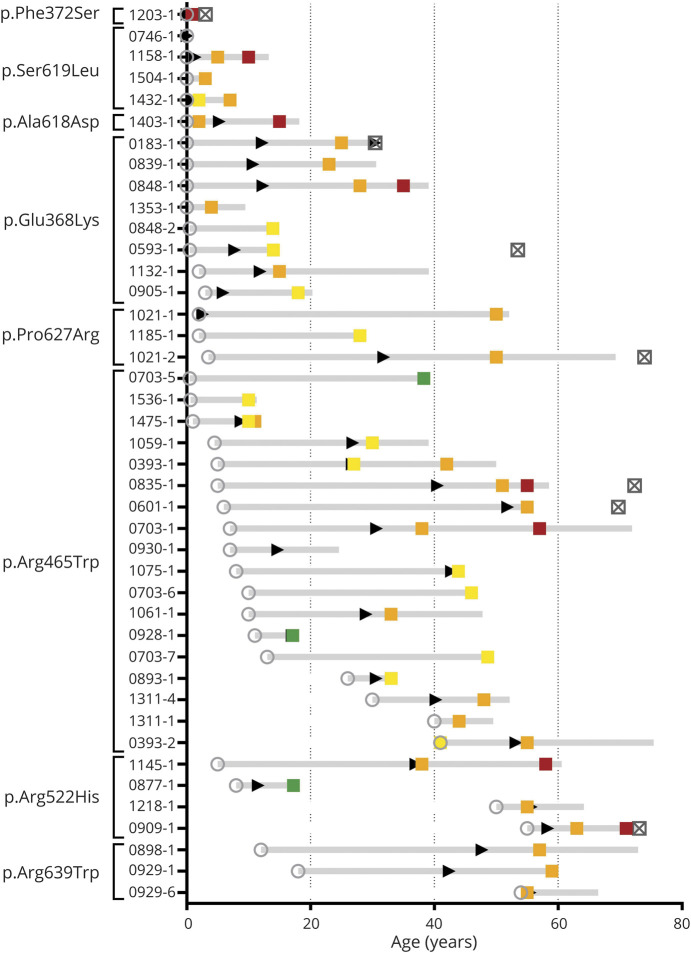
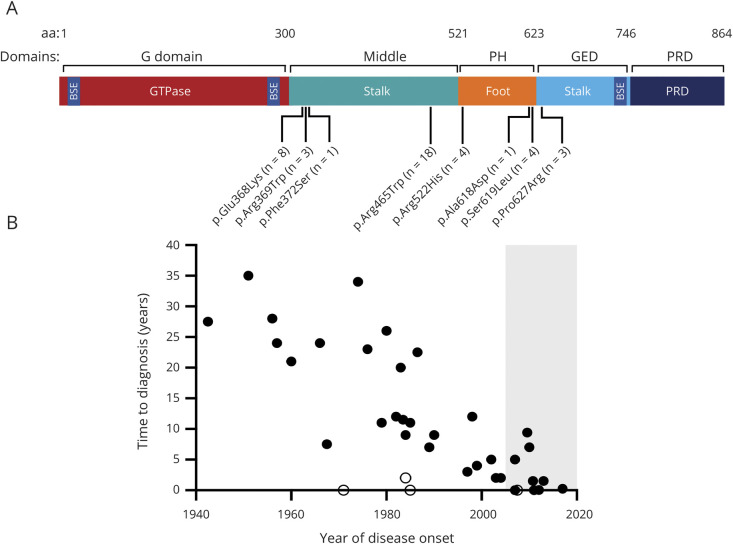
Results: Forty-two patients with DNM2-related CNM, whose ages ranged from 0.95 to 75.76 years at most recent contact, were enrolled from 34 families in North or South America and Europe. There were 8 different DNM2 pathogenic variants within the cohort. Of the 32 biopsied patients, all had histologic features of CNM. The disease onset was in infancy or childhood in 81% of the cohort, and more than half of the patients had high arched palates, indicative of weakness in utero. Ambulation was affected in nearly all (92%) the patients, and while the rapidity of progression was variable, most (67%) reported a "deteriorating course." Ptosis, ophthalmoparesis, facial weakness, dysphagia, and respiratory insufficiency were commonly reported. One-third of the patients experienced restricted jaw mobility. Certain pathogenic variants appear to correlate with a more severe phenotype.
Discussion: DNM2-related CNM has a predominantly early-onset, often congenital, myopathy resulting in progressive difficulty with ambulation and occasionally bulbar and respiratory dysfunction. This detailed characterization of the phenotype provides important information to support clinical trial readiness for future disease-modifying therapies.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


