Samantha B Bremner, Christian J Mandrycky, Andrea Leonard, Ruby M Padgett, Alan R Levinson, Ethan S Rehn, J Manuel Pioner, Nathan J Sniadecki, David L Mack
{"title":"全长肌营养不良蛋白缺乏会导致人体工程心脏组织出现收缩和钙瞬态缺陷。","authors":"Samantha B Bremner, Christian J Mandrycky, Andrea Leonard, Ruby M Padgett, Alan R Levinson, Ethan S Rehn, J Manuel Pioner, Nathan J Sniadecki, David L Mack","doi":"10.1177/20417314221119628","DOIUrl":null,"url":null,"abstract":"<p><p>Cardiomyopathy is currently the leading cause of death for patients with Duchenne muscular dystrophy (DMD), a severe neuromuscular disorder affecting young boys. Animal models have provided insight into the mechanisms by which dystrophin protein deficiency causes cardiomyopathy, but there remains a need to develop human models of DMD to validate pathogenic mechanisms and identify therapeutic targets. Here, we have developed human engineered heart tissues (EHTs) from CRISPR-edited, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) expressing a truncated dystrophin protein lacking part of the actin-binding domain. The 3D EHT platform enables direct measurement of contractile force, simultaneous monitoring of Ca<sup>2+</sup> transients, and assessment of myofibril structure. Dystrophin-mutant EHTs produced less contractile force as well as delayed kinetics of force generation and relaxation, as compared to isogenic controls. Contractile dysfunction was accompanied by reduced sarcomere length, increased resting cytosolic Ca<sup>2+</sup> levels, delayed Ca<sup>2+</sup> release and reuptake, and increased beat rate irregularity. Transcriptomic analysis revealed clear differences between dystrophin-deficient and control EHTs, including downregulation of genes related to Ca<sup>2+</sup> homeostasis and extracellular matrix organization, and upregulation of genes related to regulation of membrane potential, cardiac muscle development, and heart contraction. These findings indicate that the EHT platform provides the cues necessary to expose the clinically-relevant, functional phenotype of force production as well as mechanistic insights into the role of Ca<sup>2+</sup> handling and transcriptomic dysregulation in dystrophic cardiac function, ultimately providing a powerful platform for further studies in disease modeling and drug discovery.</p>","PeriodicalId":17384,"journal":{"name":"Journal of Tissue Engineering","volume":"13 ","pages":"20417314221119628"},"PeriodicalIF":6.7000,"publicationDate":"2022-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/58/10.1177_20417314221119628.PMC9393922.pdf","citationCount":"0","resultStr":"{\"title\":\"Full-length dystrophin deficiency leads to contractile and calcium transient defects in human engineered heart tissues.\",\"authors\":\"Samantha B Bremner, Christian J Mandrycky, Andrea Leonard, Ruby M Padgett, Alan R Levinson, Ethan S Rehn, J Manuel Pioner, Nathan J Sniadecki, David L Mack\",\"doi\":\"10.1177/20417314221119628\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cardiomyopathy is currently the leading cause of death for patients with Duchenne muscular dystrophy (DMD), a severe neuromuscular disorder affecting young boys. Animal models have provided insight into the mechanisms by which dystrophin protein deficiency causes cardiomyopathy, but there remains a need to develop human models of DMD to validate pathogenic mechanisms and identify therapeutic targets. Here, we have developed human engineered heart tissues (EHTs) from CRISPR-edited, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) expressing a truncated dystrophin protein lacking part of the actin-binding domain. The 3D EHT platform enables direct measurement of contractile force, simultaneous monitoring of Ca<sup>2+</sup> transients, and assessment of myofibril structure. Dystrophin-mutant EHTs produced less contractile force as well as delayed kinetics of force generation and relaxation, as compared to isogenic controls. Contractile dysfunction was accompanied by reduced sarcomere length, increased resting cytosolic Ca<sup>2+</sup> levels, delayed Ca<sup>2+</sup> release and reuptake, and increased beat rate irregularity. Transcriptomic analysis revealed clear differences between dystrophin-deficient and control EHTs, including downregulation of genes related to Ca<sup>2+</sup> homeostasis and extracellular matrix organization, and upregulation of genes related to regulation of membrane potential, cardiac muscle development, and heart contraction. These findings indicate that the EHT platform provides the cues necessary to expose the clinically-relevant, functional phenotype of force production as well as mechanistic insights into the role of Ca<sup>2+</sup> handling and transcriptomic dysregulation in dystrophic cardiac function, ultimately providing a powerful platform for further studies in disease modeling and drug discovery.</p>\",\"PeriodicalId\":17384,\"journal\":{\"name\":\"Journal of Tissue Engineering\",\"volume\":\"13 \",\"pages\":\"20417314221119628\"},\"PeriodicalIF\":6.7000,\"publicationDate\":\"2022-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b1/58/10.1177_20417314221119628.PMC9393922.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Tissue Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://doi.org/10.1177/20417314221119628\",\"RegionNum\":1,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"CELL & TISSUE ENGINEERING\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Tissue Engineering","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.1177/20417314221119628","RegionNum":1,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"CELL & TISSUE ENGINEERING","Score":null,"Total":0}
Full-length dystrophin deficiency leads to contractile and calcium transient defects in human engineered heart tissues.
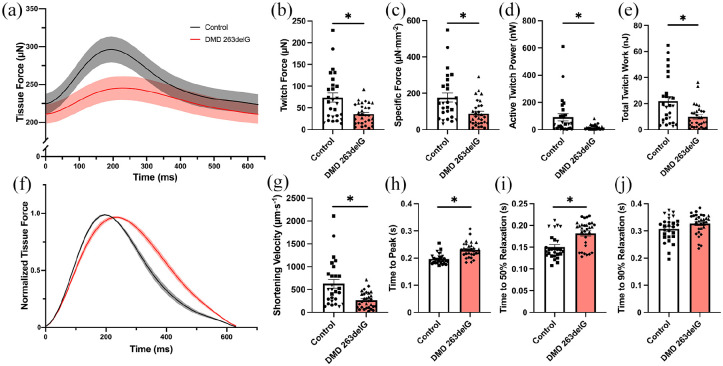
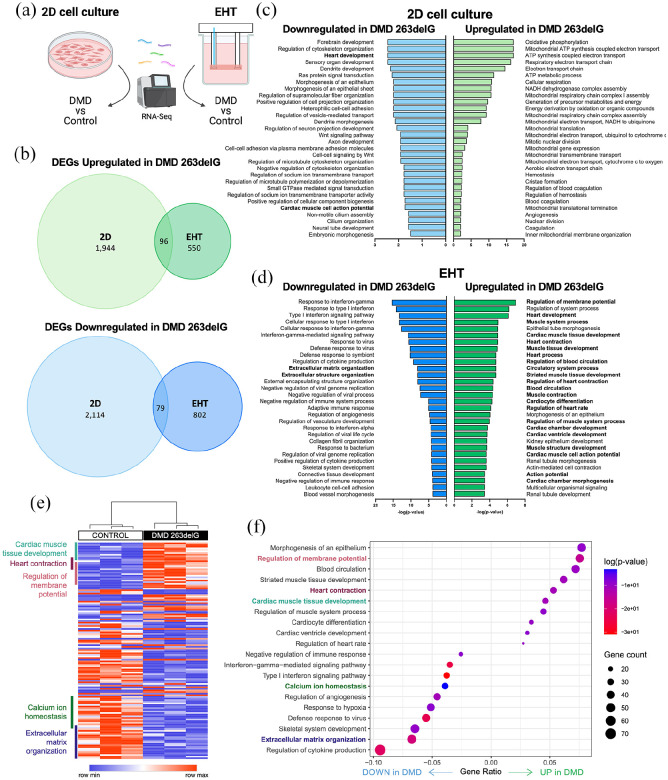
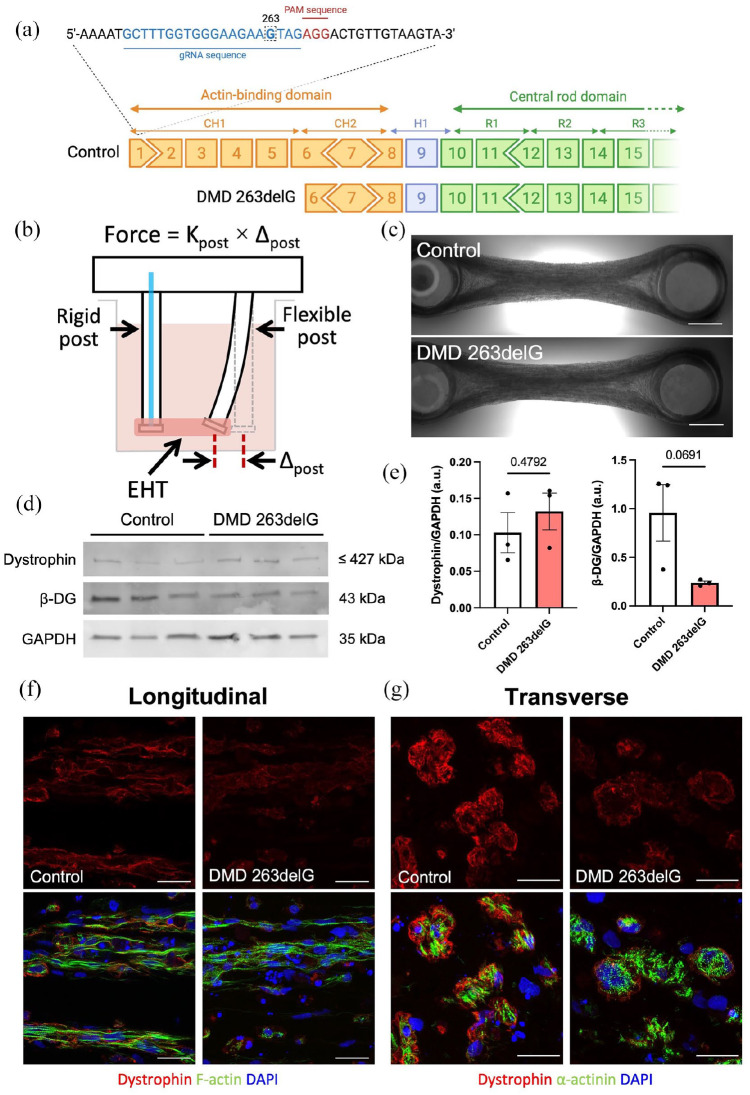
Cardiomyopathy is currently the leading cause of death for patients with Duchenne muscular dystrophy (DMD), a severe neuromuscular disorder affecting young boys. Animal models have provided insight into the mechanisms by which dystrophin protein deficiency causes cardiomyopathy, but there remains a need to develop human models of DMD to validate pathogenic mechanisms and identify therapeutic targets. Here, we have developed human engineered heart tissues (EHTs) from CRISPR-edited, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) expressing a truncated dystrophin protein lacking part of the actin-binding domain. The 3D EHT platform enables direct measurement of contractile force, simultaneous monitoring of Ca2+ transients, and assessment of myofibril structure. Dystrophin-mutant EHTs produced less contractile force as well as delayed kinetics of force generation and relaxation, as compared to isogenic controls. Contractile dysfunction was accompanied by reduced sarcomere length, increased resting cytosolic Ca2+ levels, delayed Ca2+ release and reuptake, and increased beat rate irregularity. Transcriptomic analysis revealed clear differences between dystrophin-deficient and control EHTs, including downregulation of genes related to Ca2+ homeostasis and extracellular matrix organization, and upregulation of genes related to regulation of membrane potential, cardiac muscle development, and heart contraction. These findings indicate that the EHT platform provides the cues necessary to expose the clinically-relevant, functional phenotype of force production as well as mechanistic insights into the role of Ca2+ handling and transcriptomic dysregulation in dystrophic cardiac function, ultimately providing a powerful platform for further studies in disease modeling and drug discovery.
期刊介绍:
The Journal of Tissue Engineering (JTE) is a peer-reviewed, open-access journal dedicated to scientific research in the field of tissue engineering and its clinical applications. Our journal encompasses a wide range of interests, from the fundamental aspects of stem cells and progenitor cells, including their expansion to viable numbers, to an in-depth understanding of their differentiation processes. Join us in exploring the latest advancements in tissue engineering and its clinical translation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


