Juan Carlos Rivera-Mulia, Claudia Trevilla-Garcia, Santiago Martinez-Cifuentes
{"title":"优化的Repli-seq:通过下一代测序改进DNA复制时间分析。","authors":"Juan Carlos Rivera-Mulia, Claudia Trevilla-Garcia, Santiago Martinez-Cifuentes","doi":"10.1007/s10577-022-09703-7","DOIUrl":null,"url":null,"abstract":"<p><p>The human genome is divided into functional units that replicate at specific times during S-phase. This temporal program is known as replication timing (RT) and is coordinated with the spatial organization of the genome and transcriptional activity. RT is also cell type-specific, dynamically regulated during development, and alterations in RT are observed in multiple diseases. Thus, the precise measure of RT is critical to understand the role of RT in gene function regulation. Distinct methods for assaying the RT program exist; however, conventional methods require thousands of cells as input, prohibiting its applicability to samples with limited cell numbers such as those from disease patients or from early developing embryos. Although single-cell RT analyses have been developed, these methods are low throughput, require generation of numerous libraries, increased sequencing costs, and produce low resolution data. Here, we developed an improved method to measure RT genome-wide that enables high-resolution analysis of low input samples. This method incorporates direct cell sorting into lysis buffer, as well as DNA fragmentation and library preparation in a single tube, resulting in higher yields, increased quality, and reproducibility with decreased costs. We also performed a systematic data processing analysis to provide standardized parameters for RT measurement. This optimized method facilitates RT analysis and will enable its application to a broad range of studies investigating the role of RT in gene expression, nuclear architecture, and disease.</p>","PeriodicalId":50698,"journal":{"name":"Chromosome Research","volume":"30 4","pages":"401-414"},"PeriodicalIF":2.4000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10124313/pdf/","citationCount":"0","resultStr":"{\"title\":\"Optimized Repli-seq: improved DNA replication timing analysis by next-generation sequencing.\",\"authors\":\"Juan Carlos Rivera-Mulia, Claudia Trevilla-Garcia, Santiago Martinez-Cifuentes\",\"doi\":\"10.1007/s10577-022-09703-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The human genome is divided into functional units that replicate at specific times during S-phase. This temporal program is known as replication timing (RT) and is coordinated with the spatial organization of the genome and transcriptional activity. RT is also cell type-specific, dynamically regulated during development, and alterations in RT are observed in multiple diseases. Thus, the precise measure of RT is critical to understand the role of RT in gene function regulation. Distinct methods for assaying the RT program exist; however, conventional methods require thousands of cells as input, prohibiting its applicability to samples with limited cell numbers such as those from disease patients or from early developing embryos. Although single-cell RT analyses have been developed, these methods are low throughput, require generation of numerous libraries, increased sequencing costs, and produce low resolution data. Here, we developed an improved method to measure RT genome-wide that enables high-resolution analysis of low input samples. This method incorporates direct cell sorting into lysis buffer, as well as DNA fragmentation and library preparation in a single tube, resulting in higher yields, increased quality, and reproducibility with decreased costs. We also performed a systematic data processing analysis to provide standardized parameters for RT measurement. This optimized method facilitates RT analysis and will enable its application to a broad range of studies investigating the role of RT in gene expression, nuclear architecture, and disease.</p>\",\"PeriodicalId\":50698,\"journal\":{\"name\":\"Chromosome Research\",\"volume\":\"30 4\",\"pages\":\"401-414\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10124313/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chromosome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s10577-022-09703-7\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/7/4 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chromosome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s10577-022-09703-7","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/7/4 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Optimized Repli-seq: improved DNA replication timing analysis by next-generation sequencing.
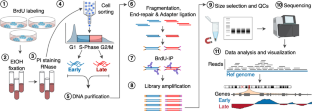
The human genome is divided into functional units that replicate at specific times during S-phase. This temporal program is known as replication timing (RT) and is coordinated with the spatial organization of the genome and transcriptional activity. RT is also cell type-specific, dynamically regulated during development, and alterations in RT are observed in multiple diseases. Thus, the precise measure of RT is critical to understand the role of RT in gene function regulation. Distinct methods for assaying the RT program exist; however, conventional methods require thousands of cells as input, prohibiting its applicability to samples with limited cell numbers such as those from disease patients or from early developing embryos. Although single-cell RT analyses have been developed, these methods are low throughput, require generation of numerous libraries, increased sequencing costs, and produce low resolution data. Here, we developed an improved method to measure RT genome-wide that enables high-resolution analysis of low input samples. This method incorporates direct cell sorting into lysis buffer, as well as DNA fragmentation and library preparation in a single tube, resulting in higher yields, increased quality, and reproducibility with decreased costs. We also performed a systematic data processing analysis to provide standardized parameters for RT measurement. This optimized method facilitates RT analysis and will enable its application to a broad range of studies investigating the role of RT in gene expression, nuclear architecture, and disease.
期刊介绍:
Chromosome Research publishes manuscripts from work based on all organisms and encourages submissions in the following areas including, but not limited, to:
· Chromosomes and their linkage to diseases;
· Chromosome organization within the nucleus;
· Chromatin biology (transcription, non-coding RNA, etc);
· Chromosome structure, function and mechanics;
· Chromosome and DNA repair;
· Epigenetic chromosomal functions (centromeres, telomeres, replication, imprinting,
dosage compensation, sex determination, chromosome remodeling);
· Architectural/epigenomic organization of the genome;
· Functional annotation of the genome;
· Functional and comparative genomics in plants and animals;
· Karyology studies that help resolve difficult taxonomic problems or that provide
clues to fundamental mechanisms of genome and karyotype evolution in plants and animals;
· Mitosis and Meiosis;
· Cancer cytogenomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


