Annika van Hummel, Miheer Sabale, Magdalena Przybyla, Julia van der Hoven, Gabriella Chan, Astrid F Feiten, Roger S Chung, Lars M Ittner, Yazi D Ke
{"title":"野生型和 ALS/FTD 突变细胞周期蛋白 F 小鼠模型中的 TDP-43 病理学和功能缺陷。","authors":"Annika van Hummel, Miheer Sabale, Magdalena Przybyla, Julia van der Hoven, Gabriella Chan, Astrid F Feiten, Roger S Chung, Lars M Ittner, Yazi D Ke","doi":"10.1111/nan.12902","DOIUrl":null,"url":null,"abstract":"<p><strong>Aims: </strong>Amyotrophic lateral sclerosis (ALS) is characterised by a progressive loss of upper and lower motor neurons leading to muscle weakness and eventually death. Frontotemporal dementia (FTD) presents clinically with significant behavioural decline. Approximately 10% of cases have a known family history, and disease-linked mutations in multiple genes have been identified in FTD and ALS. More recently, ALS and FTD-linked variants have been identified in the CCNF gene, which accounts for an estimated 0.6% to over 3% of familial ALS cases.</p><p><strong>Methods: </strong>In this study, we developed the first mouse models expressing either wild-type (WT) human CCNF or its mutant pathogenic variant S621G to recapitulate key clinical and neuropathological features of ALS and FTD linked to CCNF disease variants. We expressed human CCNF WT or CCNF<sup>S621G</sup> throughout the murine brain by intracranial delivery of adeno-associated virus (AAV) to achieve widespread delivery via somatic brain transgenesis.</p><p><strong>Results: </strong>These mice developed behavioural abnormalities, similar to the clinical symptoms of FTD patients, as early as 3 months of age, including hyperactivity and disinhibition, which progressively deteriorated to include memory deficits by 8 months of age. Brains of mutant CCNF_S621G mice displayed an accumulation of ubiquitinated proteins with elevated levels of phosphorylated TDP-43 present in both CCNF_WT and mutant CCNF_S621G mice. We also investigated the effects of CCNF expression on interaction targets of CCNF and found elevated levels of insoluble splicing factor proline and glutamine-rich (SFPQ). Furthermore, cytoplasmic TDP-43 inclusions were found in both CCNF_WT and mutant CCNF_S621G mice, recapitulating the key hallmark of FTD/ALS pathology.</p><p><strong>Conclusions: </strong>In summary, CCNF expression in mice reproduces clinical presentations of ALS, including functional deficits and TDP-43 neuropathology with altered CCNF-mediated pathways contributing to the pathology observed.</p>","PeriodicalId":19151,"journal":{"name":"Neuropathology and Applied Neurobiology","volume":"49 2","pages":"e12902"},"PeriodicalIF":4.0000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10946706/pdf/","citationCount":"0","resultStr":"{\"title\":\"TDP-43 pathology and functional deficits in wild-type and ALS/FTD mutant cyclin F mouse models.\",\"authors\":\"Annika van Hummel, Miheer Sabale, Magdalena Przybyla, Julia van der Hoven, Gabriella Chan, Astrid F Feiten, Roger S Chung, Lars M Ittner, Yazi D Ke\",\"doi\":\"10.1111/nan.12902\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Aims: </strong>Amyotrophic lateral sclerosis (ALS) is characterised by a progressive loss of upper and lower motor neurons leading to muscle weakness and eventually death. Frontotemporal dementia (FTD) presents clinically with significant behavioural decline. Approximately 10% of cases have a known family history, and disease-linked mutations in multiple genes have been identified in FTD and ALS. More recently, ALS and FTD-linked variants have been identified in the CCNF gene, which accounts for an estimated 0.6% to over 3% of familial ALS cases.</p><p><strong>Methods: </strong>In this study, we developed the first mouse models expressing either wild-type (WT) human CCNF or its mutant pathogenic variant S621G to recapitulate key clinical and neuropathological features of ALS and FTD linked to CCNF disease variants. We expressed human CCNF WT or CCNF<sup>S621G</sup> throughout the murine brain by intracranial delivery of adeno-associated virus (AAV) to achieve widespread delivery via somatic brain transgenesis.</p><p><strong>Results: </strong>These mice developed behavioural abnormalities, similar to the clinical symptoms of FTD patients, as early as 3 months of age, including hyperactivity and disinhibition, which progressively deteriorated to include memory deficits by 8 months of age. Brains of mutant CCNF_S621G mice displayed an accumulation of ubiquitinated proteins with elevated levels of phosphorylated TDP-43 present in both CCNF_WT and mutant CCNF_S621G mice. We also investigated the effects of CCNF expression on interaction targets of CCNF and found elevated levels of insoluble splicing factor proline and glutamine-rich (SFPQ). Furthermore, cytoplasmic TDP-43 inclusions were found in both CCNF_WT and mutant CCNF_S621G mice, recapitulating the key hallmark of FTD/ALS pathology.</p><p><strong>Conclusions: </strong>In summary, CCNF expression in mice reproduces clinical presentations of ALS, including functional deficits and TDP-43 neuropathology with altered CCNF-mediated pathways contributing to the pathology observed.</p>\",\"PeriodicalId\":19151,\"journal\":{\"name\":\"Neuropathology and Applied Neurobiology\",\"volume\":\"49 2\",\"pages\":\"e12902\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2023-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10946706/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neuropathology and Applied Neurobiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1111/nan.12902\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuropathology and Applied Neurobiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1111/nan.12902","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
目的:肌萎缩侧索硬化症(ALS)的特征是上下运动神经元逐渐丧失,导致肌肉无力,最终死亡。额颞叶痴呆症(FTD)临床表现为明显的行为衰退。约 10% 的病例有已知的家族史,在 FTD 和 ALS 中发现了多个与疾病相关的基因突变。最近,在 CCNF 基因中发现了与 ALS 和 FTD 相关的变异,据估计,家族性 ALS 病例中 0.6% 至 3% 以上是由 CCNF 基因引起的:在这项研究中,我们首次开发了表达野生型(WT)人类CCNF或其突变致病变体S621G的小鼠模型,以再现与CCNF疾病变体相关的ALS和FTD的主要临床和神经病理学特征。我们通过颅内注射腺相关病毒(AAV)在整个小鼠大脑中表达了人CCNF WT或CCNFS621G,从而通过体细胞脑转基因技术实现了广泛注射:这些小鼠早在3月龄时就出现了行为异常,与FTD患者的临床症状相似,包括多动和抑制,并在8月龄时逐渐恶化为记忆障碍。突变型CCNF_S621G小鼠大脑中泛素化蛋白质堆积,磷酸化TDP-43水平升高,CCNF_WT和突变型CCNF_S621G小鼠均存在这种情况。我们还研究了CCNF表达对CCNF相互作用靶标的影响,发现不溶性剪接因子富脯氨酸和谷氨酰胺(SFPQ)水平升高。此外,在CCNF_WT和突变体CCNF_S621G小鼠中都发现了细胞质TDP-43包涵体,再现了FTD/ALS病理学的关键特征:总之,CCNF在小鼠中的表达再现了ALS的临床表现,包括功能障碍和TDP-43神经病理学,CCNF介导的途径改变导致了所观察到的病理学。
TDP-43 pathology and functional deficits in wild-type and ALS/FTD mutant cyclin F mouse models.
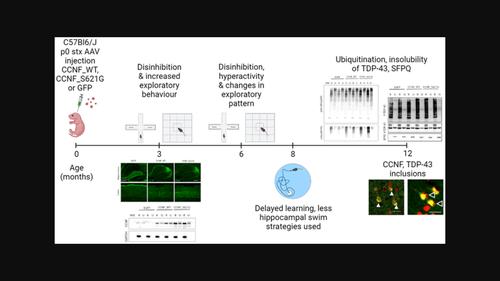
Aims: Amyotrophic lateral sclerosis (ALS) is characterised by a progressive loss of upper and lower motor neurons leading to muscle weakness and eventually death. Frontotemporal dementia (FTD) presents clinically with significant behavioural decline. Approximately 10% of cases have a known family history, and disease-linked mutations in multiple genes have been identified in FTD and ALS. More recently, ALS and FTD-linked variants have been identified in the CCNF gene, which accounts for an estimated 0.6% to over 3% of familial ALS cases.
Methods: In this study, we developed the first mouse models expressing either wild-type (WT) human CCNF or its mutant pathogenic variant S621G to recapitulate key clinical and neuropathological features of ALS and FTD linked to CCNF disease variants. We expressed human CCNF WT or CCNFS621G throughout the murine brain by intracranial delivery of adeno-associated virus (AAV) to achieve widespread delivery via somatic brain transgenesis.
Results: These mice developed behavioural abnormalities, similar to the clinical symptoms of FTD patients, as early as 3 months of age, including hyperactivity and disinhibition, which progressively deteriorated to include memory deficits by 8 months of age. Brains of mutant CCNF_S621G mice displayed an accumulation of ubiquitinated proteins with elevated levels of phosphorylated TDP-43 present in both CCNF_WT and mutant CCNF_S621G mice. We also investigated the effects of CCNF expression on interaction targets of CCNF and found elevated levels of insoluble splicing factor proline and glutamine-rich (SFPQ). Furthermore, cytoplasmic TDP-43 inclusions were found in both CCNF_WT and mutant CCNF_S621G mice, recapitulating the key hallmark of FTD/ALS pathology.
Conclusions: In summary, CCNF expression in mice reproduces clinical presentations of ALS, including functional deficits and TDP-43 neuropathology with altered CCNF-mediated pathways contributing to the pathology observed.
期刊介绍:
Neuropathology and Applied Neurobiology is an international journal for the publication of original papers, both clinical and experimental, on problems and pathological processes in neuropathology and muscle disease. Established in 1974, this reputable and well respected journal is an international journal sponsored by the British Neuropathological Society, one of the world leading societies for Neuropathology, pioneering research and scientific endeavour with a global membership base. Additionally members of the British Neuropathological Society get 50% off the cost of print colour on acceptance of their article.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


