William Pilcher, Beena E Thomas, Swati S Bhasin, Reyka G Jayasinghe, Lijun Yao, Edgar Gonzalez-Kozlova, Surendra Dasari, Seunghee Kim-Schulze, Adeeb Rahman, Jonathan Patton, Mark Fiala, Giulia Cheloni, Taxiarchis Kourelis, Madhav V Dhodapkar, Ravi Vij, Shaadi Mehr, Mark Hamilton, Hearn Jay Cho, Daniel Auclair, David E Avigan, Shaji K Kumar, Sacha Gnjatic, Li Ding, Manoj Bhasin
{"title":"对进展迅速的多发性骨髓瘤免疫微环境的跨中心单细胞 RNA 测序研究。","authors":"William Pilcher, Beena E Thomas, Swati S Bhasin, Reyka G Jayasinghe, Lijun Yao, Edgar Gonzalez-Kozlova, Surendra Dasari, Seunghee Kim-Schulze, Adeeb Rahman, Jonathan Patton, Mark Fiala, Giulia Cheloni, Taxiarchis Kourelis, Madhav V Dhodapkar, Ravi Vij, Shaadi Mehr, Mark Hamilton, Hearn Jay Cho, Daniel Auclair, David E Avigan, Shaji K Kumar, Sacha Gnjatic, Li Ding, Manoj Bhasin","doi":"10.1038/s41525-022-00340-x","DOIUrl":null,"url":null,"abstract":"<p><p>Despite advancements in understanding the pathophysiology of Multiple Myeloma (MM), the cause of rapid progressing disease in a subset of patients is still unclear. MM's progression is facilitated by complex interactions with the surrounding bone marrow (BM) cells, forming a microenvironment that supports tumor growth and drug resistance. Understanding the immune microenvironment is key to identifying factors that promote rapid progression of MM. To accomplish this, we performed a multi-center single-cell RNA sequencing (scRNA-seq) study on 102,207 cells from 48 CD138<sup>-</sup> BM samples collected at the time of disease diagnosis from 18 patients with either rapid progressing (progression-free survival (PFS) < 18 months) or non-progressing (PFS > 4 years) disease. Comparative analysis of data from three centers demonstrated similar transcriptome profiles and cell type distributions, indicating subtle technical variation in scRNA-seq, opening avenues for an expanded multicenter trial. Rapid progressors depicted significantly higher enrichment of GZMK<sup>+</sup> and TIGIT<sup>+</sup> exhausted CD8<sup>+</sup> T-cells (P = 0.022) along with decreased expression of cytolytic markers (PRF1, GZMB, GNLY). We also observed a significantly higher enrichment of M2 tolerogenic macrophages in rapid progressors and activation of pro-proliferative signaling pathways, such as BAFF, CCL, and IL16. On the other hand, non-progressive patients depicted higher enrichment for immature B Cells (i.e., Pre/Pro B cells), with elevated expression for markers of B cell development (IGLL1, SOX4, DNTT). This multi-center study identifies the enrichment of various pro-tumorigenic cell populations and pathways in those with rapid progressing disease and further validates the robustness of scRNA-seq data generated at different study centers.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"8 1","pages":"3"},"PeriodicalIF":4.7000,"publicationDate":"2023-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9879959/pdf/","citationCount":"0","resultStr":"{\"title\":\"Cross center single-cell RNA sequencing study of the immune microenvironment in rapid progressing multiple myeloma.\",\"authors\":\"William Pilcher, Beena E Thomas, Swati S Bhasin, Reyka G Jayasinghe, Lijun Yao, Edgar Gonzalez-Kozlova, Surendra Dasari, Seunghee Kim-Schulze, Adeeb Rahman, Jonathan Patton, Mark Fiala, Giulia Cheloni, Taxiarchis Kourelis, Madhav V Dhodapkar, Ravi Vij, Shaadi Mehr, Mark Hamilton, Hearn Jay Cho, Daniel Auclair, David E Avigan, Shaji K Kumar, Sacha Gnjatic, Li Ding, Manoj Bhasin\",\"doi\":\"10.1038/s41525-022-00340-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Despite advancements in understanding the pathophysiology of Multiple Myeloma (MM), the cause of rapid progressing disease in a subset of patients is still unclear. MM's progression is facilitated by complex interactions with the surrounding bone marrow (BM) cells, forming a microenvironment that supports tumor growth and drug resistance. Understanding the immune microenvironment is key to identifying factors that promote rapid progression of MM. To accomplish this, we performed a multi-center single-cell RNA sequencing (scRNA-seq) study on 102,207 cells from 48 CD138<sup>-</sup> BM samples collected at the time of disease diagnosis from 18 patients with either rapid progressing (progression-free survival (PFS) < 18 months) or non-progressing (PFS > 4 years) disease. Comparative analysis of data from three centers demonstrated similar transcriptome profiles and cell type distributions, indicating subtle technical variation in scRNA-seq, opening avenues for an expanded multicenter trial. Rapid progressors depicted significantly higher enrichment of GZMK<sup>+</sup> and TIGIT<sup>+</sup> exhausted CD8<sup>+</sup> T-cells (P = 0.022) along with decreased expression of cytolytic markers (PRF1, GZMB, GNLY). We also observed a significantly higher enrichment of M2 tolerogenic macrophages in rapid progressors and activation of pro-proliferative signaling pathways, such as BAFF, CCL, and IL16. On the other hand, non-progressive patients depicted higher enrichment for immature B Cells (i.e., Pre/Pro B cells), with elevated expression for markers of B cell development (IGLL1, SOX4, DNTT). This multi-center study identifies the enrichment of various pro-tumorigenic cell populations and pathways in those with rapid progressing disease and further validates the robustness of scRNA-seq data generated at different study centers.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"8 1\",\"pages\":\"3\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2023-01-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9879959/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-022-00340-x\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-022-00340-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
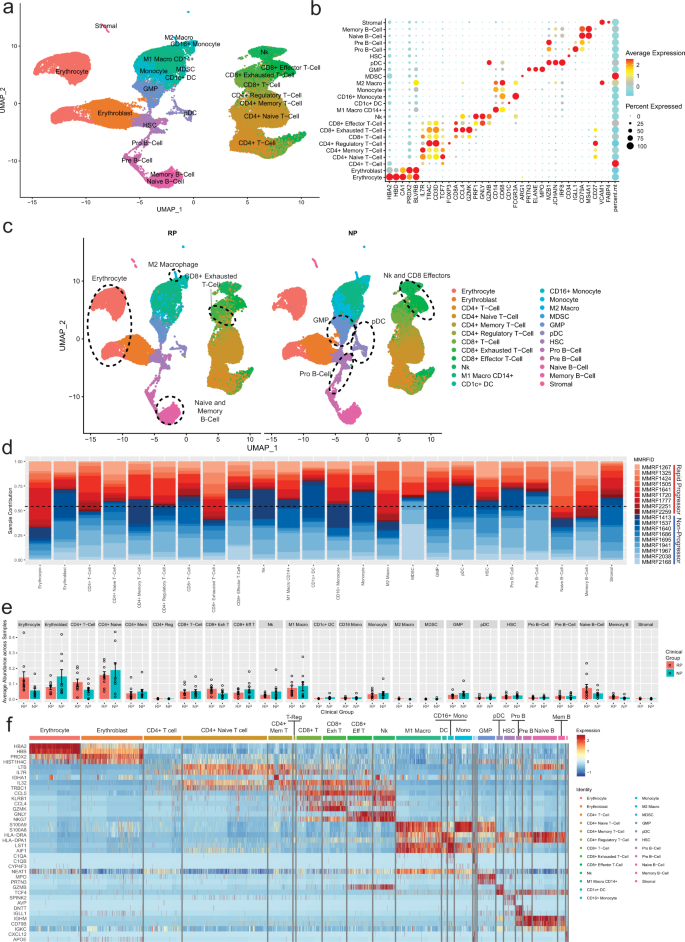
尽管人们对多发性骨髓瘤(MM)的病理生理学有了进一步的了解,但仍不清楚导致部分患者病情快速进展的原因。多发性骨髓瘤与周围骨髓(BM)细胞的复杂相互作用促进了病情的进展,形成了支持肿瘤生长和耐药性的微环境。了解免疫微环境是确定促进 MM 快速进展的因素的关键。为了实现这一目标,我们进行了一项多中心单细胞RNA测序(scRNA-seq)研究,研究对象是18名快速进展期(无进展生存期(PFS)4年)患者在疾病诊断时收集的48份CD138-BM样本中的102207个细胞。对来自三个中心的数据进行的比较分析表明,转录组图谱和细胞类型分布相似,表明scRNA-seq技术存在细微差异,为扩大多中心试验开辟了途径。快速进展者的 CD8+ T 细胞中 GZMK+ 和 TIGIT+ 的富集度明显更高(P = 0.022),同时细胞溶解标志物(PRF1、GZMB、GNLY)的表达也有所下降。我们还观察到,在快速进展者中,M2耐受性巨噬细胞的富集程度明显更高,而且促增殖信号通路(如 BAFF、CCL 和 IL16)也被激活。另一方面,非进展期患者的未成熟 B 细胞(即 Pre/Pro B 细胞)富集度较高,B 细胞发育标志物(IGLL1、SOX4、DNTT)表达升高。这项多中心研究确定了各种促肿瘤细胞群和通路在疾病快速进展患者中的富集情况,并进一步验证了不同研究中心生成的 scRNA-seq 数据的稳健性。
Cross center single-cell RNA sequencing study of the immune microenvironment in rapid progressing multiple myeloma.
Despite advancements in understanding the pathophysiology of Multiple Myeloma (MM), the cause of rapid progressing disease in a subset of patients is still unclear. MM's progression is facilitated by complex interactions with the surrounding bone marrow (BM) cells, forming a microenvironment that supports tumor growth and drug resistance. Understanding the immune microenvironment is key to identifying factors that promote rapid progression of MM. To accomplish this, we performed a multi-center single-cell RNA sequencing (scRNA-seq) study on 102,207 cells from 48 CD138- BM samples collected at the time of disease diagnosis from 18 patients with either rapid progressing (progression-free survival (PFS) < 18 months) or non-progressing (PFS > 4 years) disease. Comparative analysis of data from three centers demonstrated similar transcriptome profiles and cell type distributions, indicating subtle technical variation in scRNA-seq, opening avenues for an expanded multicenter trial. Rapid progressors depicted significantly higher enrichment of GZMK+ and TIGIT+ exhausted CD8+ T-cells (P = 0.022) along with decreased expression of cytolytic markers (PRF1, GZMB, GNLY). We also observed a significantly higher enrichment of M2 tolerogenic macrophages in rapid progressors and activation of pro-proliferative signaling pathways, such as BAFF, CCL, and IL16. On the other hand, non-progressive patients depicted higher enrichment for immature B Cells (i.e., Pre/Pro B cells), with elevated expression for markers of B cell development (IGLL1, SOX4, DNTT). This multi-center study identifies the enrichment of various pro-tumorigenic cell populations and pathways in those with rapid progressing disease and further validates the robustness of scRNA-seq data generated at different study centers.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


