Jinan Wang, Hung N. Do, Kushal Koirala and Yinglong Miao*,
{"title":"预测生物分子结合动力学:综述","authors":"Jinan Wang, Hung N. Do, Kushal Koirala and Yinglong Miao*, ","doi":"10.1021/acs.jctc.2c01085","DOIUrl":null,"url":null,"abstract":"<p >Biomolecular binding kinetics including the association (<i>k</i><sub><i>on</i></sub>) and dissociation (<i>k</i><sub><i>off</i></sub>) rates are critical parameters for therapeutic design of small-molecule drugs, peptides, and antibodies. Notably, the drug molecule residence time or dissociation rate has been shown to correlate with their efficacies better than binding affinities. A wide range of modeling approaches including quantitative structure-kinetic relationship models, Molecular Dynamics simulations, enhanced sampling, and Machine Learning has been developed to explore biomolecular binding and dissociation mechanisms and predict binding kinetic rates. Here, we review recent advances in computational modeling of biomolecular binding kinetics, with an outlook for future improvements.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"19 8","pages":"2135–2148"},"PeriodicalIF":5.7000,"publicationDate":"2023-03-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"7","resultStr":"{\"title\":\"Predicting Biomolecular Binding Kinetics: A Review\",\"authors\":\"Jinan Wang, Hung N. Do, Kushal Koirala and Yinglong Miao*, \",\"doi\":\"10.1021/acs.jctc.2c01085\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Biomolecular binding kinetics including the association (<i>k</i><sub><i>on</i></sub>) and dissociation (<i>k</i><sub><i>off</i></sub>) rates are critical parameters for therapeutic design of small-molecule drugs, peptides, and antibodies. Notably, the drug molecule residence time or dissociation rate has been shown to correlate with their efficacies better than binding affinities. A wide range of modeling approaches including quantitative structure-kinetic relationship models, Molecular Dynamics simulations, enhanced sampling, and Machine Learning has been developed to explore biomolecular binding and dissociation mechanisms and predict binding kinetic rates. Here, we review recent advances in computational modeling of biomolecular binding kinetics, with an outlook for future improvements.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"19 8\",\"pages\":\"2135–2148\"},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2023-03-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.2c01085\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.2c01085","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Predicting Biomolecular Binding Kinetics: A Review
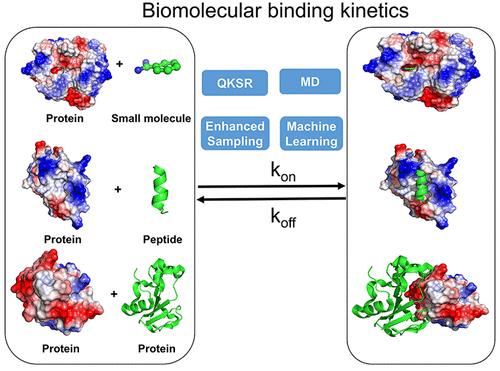
Biomolecular binding kinetics including the association (kon) and dissociation (koff) rates are critical parameters for therapeutic design of small-molecule drugs, peptides, and antibodies. Notably, the drug molecule residence time or dissociation rate has been shown to correlate with their efficacies better than binding affinities. A wide range of modeling approaches including quantitative structure-kinetic relationship models, Molecular Dynamics simulations, enhanced sampling, and Machine Learning has been developed to explore biomolecular binding and dissociation mechanisms and predict binding kinetic rates. Here, we review recent advances in computational modeling of biomolecular binding kinetics, with an outlook for future improvements.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


