{"title":"构建基于CD8+ T细胞相关基因的甲状腺癌预后风险模型。","authors":"Yaojie Hu, Xin Guo, Hong Chen, Qing Chang, Haodong Lu, Yanbing Li, Chunyou Chen","doi":"10.5114/ceji.2022.119171","DOIUrl":null,"url":null,"abstract":"<p><p>Thyroid cancer (TC) is a common and curable endocrine tumor occurring in the head and neck characterized by a low mortality rate compared to other malignancies. In this study, the immune microenvironment of TC was investigated to identify biomarkers. The mRNA and clinical data available in this study were accessed from The Cancer Genome Atlas-Thyroid Cancer (TCGA-THCA) dataset. Differences in immune infiltration levels of TC and normal samples were assessed by CIBERSORT. Thyroid cancer samples were classified into high- and low-abundance groups according to the median abundance of immune cell infiltration, and CD8<sup>+</sup> T cells were notably correlated with the survival status. Differential expression analysis was conducted on CD8<sup>+</sup> T cells to obtain immune-related differentially expressed genes (DEGs). Subsequently, a prognostic risk model was established through Cox regression analysis. According to the median risk score, samples in the training set and validation set were assigned to high- and low-risk groups. The survival and ROC curves demonstrated that the model possesses favorable prognostic prediction ability. Furthermore, the results of gene set enrichment analysis (GSEA) indicated differences between the high- and low-risk groups in terms of ECM receptor interaction and transforming growth factor β (TGF-β) signaling pathways. The tumor microenvironment of TC samples was evaluated by ESTIMATE, which showed that stromal scores were higher in the high-risk group. Finally, simple-sample GSEA (ssGSEA) was performed on TC samples. The results indicated a higher infiltration level of NK cells in the low-risk group, as well as a lower level in the high-risk group. In terms of immune function-related gene sets, genes related to APC co-inhibition, cytolytic activity, HLA and T cell co-inhibition were observed to present higher expression levels in the low-risk group. In general, this study built a 6-gene prognostic risk assessment model based on CD8<sup>+</sup> T cells through bioinformatics analysis, which is expected to be a reference for clinicians to judge the prognosis of TC patients.</p>","PeriodicalId":9694,"journal":{"name":"Central European Journal of Immunology","volume":"47 3","pages":"234-245"},"PeriodicalIF":1.5000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/2a/6b/CEJI-47-47729.PMC9896991.pdf","citationCount":"1","resultStr":"{\"title\":\"Constructing a thyroid cancer prognostic risk model based on CD8<sup>+</sup> T cell associated genes.\",\"authors\":\"Yaojie Hu, Xin Guo, Hong Chen, Qing Chang, Haodong Lu, Yanbing Li, Chunyou Chen\",\"doi\":\"10.5114/ceji.2022.119171\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Thyroid cancer (TC) is a common and curable endocrine tumor occurring in the head and neck characterized by a low mortality rate compared to other malignancies. In this study, the immune microenvironment of TC was investigated to identify biomarkers. The mRNA and clinical data available in this study were accessed from The Cancer Genome Atlas-Thyroid Cancer (TCGA-THCA) dataset. Differences in immune infiltration levels of TC and normal samples were assessed by CIBERSORT. Thyroid cancer samples were classified into high- and low-abundance groups according to the median abundance of immune cell infiltration, and CD8<sup>+</sup> T cells were notably correlated with the survival status. Differential expression analysis was conducted on CD8<sup>+</sup> T cells to obtain immune-related differentially expressed genes (DEGs). Subsequently, a prognostic risk model was established through Cox regression analysis. According to the median risk score, samples in the training set and validation set were assigned to high- and low-risk groups. The survival and ROC curves demonstrated that the model possesses favorable prognostic prediction ability. Furthermore, the results of gene set enrichment analysis (GSEA) indicated differences between the high- and low-risk groups in terms of ECM receptor interaction and transforming growth factor β (TGF-β) signaling pathways. The tumor microenvironment of TC samples was evaluated by ESTIMATE, which showed that stromal scores were higher in the high-risk group. Finally, simple-sample GSEA (ssGSEA) was performed on TC samples. The results indicated a higher infiltration level of NK cells in the low-risk group, as well as a lower level in the high-risk group. In terms of immune function-related gene sets, genes related to APC co-inhibition, cytolytic activity, HLA and T cell co-inhibition were observed to present higher expression levels in the low-risk group. In general, this study built a 6-gene prognostic risk assessment model based on CD8<sup>+</sup> T cells through bioinformatics analysis, which is expected to be a reference for clinicians to judge the prognosis of TC patients.</p>\",\"PeriodicalId\":9694,\"journal\":{\"name\":\"Central European Journal of Immunology\",\"volume\":\"47 3\",\"pages\":\"234-245\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/2a/6b/CEJI-47-47729.PMC9896991.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Central European Journal of Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.5114/ceji.2022.119171\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Central European Journal of Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.5114/ceji.2022.119171","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 1
摘要
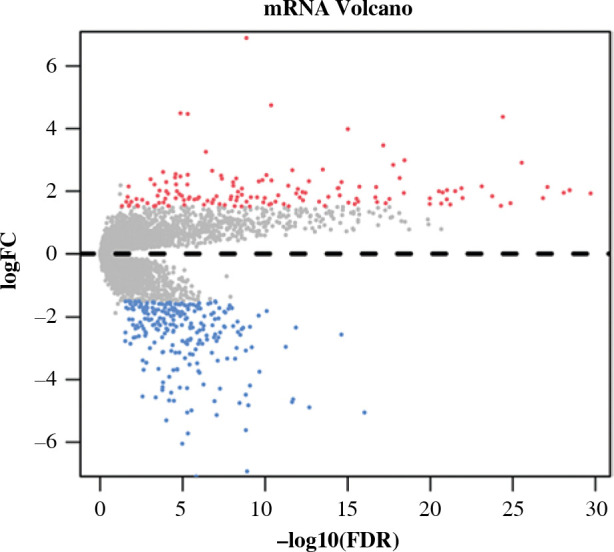
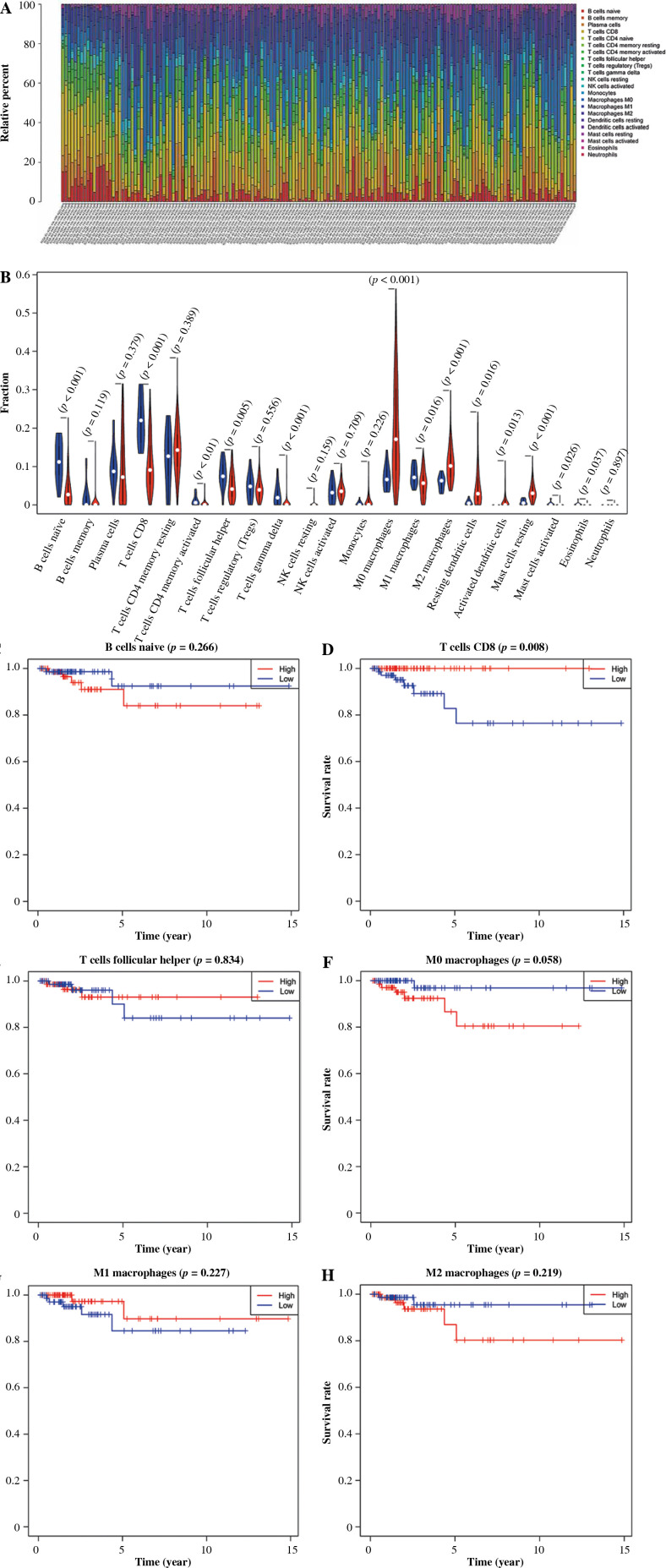
甲状腺癌(TC)是发生在头颈部的一种常见且可治愈的内分泌肿瘤,与其他恶性肿瘤相比,其死亡率较低。在本研究中,研究了TC的免疫微环境,以确定生物标志物。本研究中可用的mRNA和临床数据来自The Cancer Genome Atlas-Thyroid Cancer (TCGA-THCA)数据集。采用CIBERSORT评估TC与正常标本免疫浸润水平的差异。根据免疫细胞浸润的中位数丰度将甲状腺癌样本分为高丰度组和低丰度组,CD8+ T细胞与生存状态显著相关。对CD8+ T细胞进行差异表达分析,获得免疫相关差异表达基因(DEGs)。随后,通过Cox回归分析建立预后风险模型。根据中位风险评分,将训练集和验证集中的样本分为高危组和低危组。生存曲线和ROC曲线表明该模型具有良好的预后预测能力。此外,基因集富集分析(GSEA)结果显示,高风险组和低风险组在ECM受体相互作用和转化生长因子β (TGF-β)信号通路方面存在差异。采用ESTIMATE对TC样品的肿瘤微环境进行评估,结果显示高危组间质评分较高。最后,对TC样品进行简单样本GSEA (ssGSEA)。结果显示,低危组NK细胞浸润水平较高,高危组NK细胞浸润水平较低。免疫功能相关基因集方面,低危组APC共抑制、细胞溶解活性、HLA和T细胞共抑制相关基因表达水平较高。总体而言,本研究通过生物信息学分析,构建了基于CD8+ T细胞的6基因预后风险评估模型,有望为临床医生判断TC患者预后提供参考。
Constructing a thyroid cancer prognostic risk model based on CD8+ T cell associated genes.
Thyroid cancer (TC) is a common and curable endocrine tumor occurring in the head and neck characterized by a low mortality rate compared to other malignancies. In this study, the immune microenvironment of TC was investigated to identify biomarkers. The mRNA and clinical data available in this study were accessed from The Cancer Genome Atlas-Thyroid Cancer (TCGA-THCA) dataset. Differences in immune infiltration levels of TC and normal samples were assessed by CIBERSORT. Thyroid cancer samples were classified into high- and low-abundance groups according to the median abundance of immune cell infiltration, and CD8+ T cells were notably correlated with the survival status. Differential expression analysis was conducted on CD8+ T cells to obtain immune-related differentially expressed genes (DEGs). Subsequently, a prognostic risk model was established through Cox regression analysis. According to the median risk score, samples in the training set and validation set were assigned to high- and low-risk groups. The survival and ROC curves demonstrated that the model possesses favorable prognostic prediction ability. Furthermore, the results of gene set enrichment analysis (GSEA) indicated differences between the high- and low-risk groups in terms of ECM receptor interaction and transforming growth factor β (TGF-β) signaling pathways. The tumor microenvironment of TC samples was evaluated by ESTIMATE, which showed that stromal scores were higher in the high-risk group. Finally, simple-sample GSEA (ssGSEA) was performed on TC samples. The results indicated a higher infiltration level of NK cells in the low-risk group, as well as a lower level in the high-risk group. In terms of immune function-related gene sets, genes related to APC co-inhibition, cytolytic activity, HLA and T cell co-inhibition were observed to present higher expression levels in the low-risk group. In general, this study built a 6-gene prognostic risk assessment model based on CD8+ T cells through bioinformatics analysis, which is expected to be a reference for clinicians to judge the prognosis of TC patients.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


