Chunwang Li, Penghui Liu, Weilin Huang, Haojie Wang, Ke Ma, Lingyun Zhuo, Yaqing Kang, Qiu He, Yuanxiang Lin, Dezhi Kang, Fuxin Lin
{"title":"在中国多发性脑海绵状血管病家族中发现一种新的KRIT1/CCM1突变伴随NOTCH3突变","authors":"Chunwang Li, Penghui Liu, Weilin Huang, Haojie Wang, Ke Ma, Lingyun Zhuo, Yaqing Kang, Qiu He, Yuanxiang Lin, Dezhi Kang, Fuxin Lin","doi":"10.1007/s10048-023-00714-y","DOIUrl":null,"url":null,"abstract":"<p><p>Family cerebral cavernous malformations (FCCMs) are mainly inherited through the mutation of classical CCM genes, including CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10. FCCMs can cause severe clinical symptoms, including epileptic seizures, intracranial hemorrhage (ICH), or functional neurological deficits (FNDs). In this study, we reported a novel mutation in KRIT1 accompanied by a NOTCH3 mutation in a Chinese family. This family consists of 8 members, 4 of whom had been diagnosed with CCMs using cerebral MRI (T1WI, T2WI, SWI). The proband (II-2) and her daughter (III-4) had intracerebral hemorrhage and refractory epilepsy, respectively. Based on whole-exome sequencing (WES) data and bioinformatics analysis from 4 patients with multiple CCMs and 2 normal first-degree relatives, a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in intron 13 was considered a pathogenic gene in this family. Furthermore, based on 2 severe and 2 mild CCM patients, we found an SNV missense mutation, NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C), in NOTCH3. Finally, the KRIT1 and NOTCH3 mutations were validated in 8 members using Sanger sequencing. This study revealed a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in a Chinese CCM family, which had not been reported previously. Moreover, the NOTCH3 mutation NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C) might be a second hit and associated with the progression of CCM lesions and severe clinical symptoms.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 2","pages":"137-146"},"PeriodicalIF":1.2000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A novel KRIT1/CCM1 mutation accompanied by a NOTCH3 mutation in a Chinese family with multiple cerebral cavernous malformations.\",\"authors\":\"Chunwang Li, Penghui Liu, Weilin Huang, Haojie Wang, Ke Ma, Lingyun Zhuo, Yaqing Kang, Qiu He, Yuanxiang Lin, Dezhi Kang, Fuxin Lin\",\"doi\":\"10.1007/s10048-023-00714-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Family cerebral cavernous malformations (FCCMs) are mainly inherited through the mutation of classical CCM genes, including CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10. FCCMs can cause severe clinical symptoms, including epileptic seizures, intracranial hemorrhage (ICH), or functional neurological deficits (FNDs). In this study, we reported a novel mutation in KRIT1 accompanied by a NOTCH3 mutation in a Chinese family. This family consists of 8 members, 4 of whom had been diagnosed with CCMs using cerebral MRI (T1WI, T2WI, SWI). The proband (II-2) and her daughter (III-4) had intracerebral hemorrhage and refractory epilepsy, respectively. Based on whole-exome sequencing (WES) data and bioinformatics analysis from 4 patients with multiple CCMs and 2 normal first-degree relatives, a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in intron 13 was considered a pathogenic gene in this family. Furthermore, based on 2 severe and 2 mild CCM patients, we found an SNV missense mutation, NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C), in NOTCH3. Finally, the KRIT1 and NOTCH3 mutations were validated in 8 members using Sanger sequencing. This study revealed a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in a Chinese CCM family, which had not been reported previously. Moreover, the NOTCH3 mutation NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C) might be a second hit and associated with the progression of CCM lesions and severe clinical symptoms.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\"24 2\",\"pages\":\"137-146\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-023-00714-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00714-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
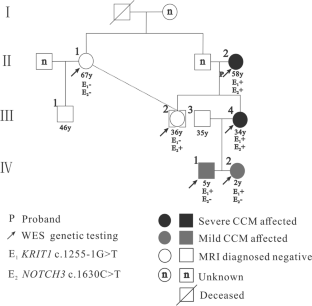
家族性脑海绵状血管瘤(FCCMs)主要通过CCM1/KRIT1、CCM2/MGC4607、CCM3/PDCD10等经典CCM基因突变进行遗传。FCCMs可引起严重的临床症状,包括癫痫发作、颅内出血(ICH)或功能性神经功能缺陷(FNDs)。在这项研究中,我们报道了一个中国家庭中KRIT1突变伴随NOTCH3突变的新突变。该家族共有8名成员,其中4人通过脑MRI (T1WI, T2WI, SWI)诊断为CCMs。先证者(II-2)及其女儿(III-4)分别有脑出血和难治性癫痫。基于4例多发性CCMs患者和2例正常一级亲属的全外显子组测序(WES)数据和生物信息学分析,13内含子中一个新的KRIT1突变NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3)被认为是该家族的致病基因。此外,在2例重度和2例轻度CCM患者中,我们在NOTCH3中发现了SNV错义突变NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C)。最后,使用Sanger测序在8个成员中验证了KRIT1和NOTCH3突变。本研究在中国CCM家族中发现了一个新的KRIT1突变NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3),该突变此前未被报道。此外,NOTCH3突变NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C)可能是第二个打击,并与CCM病变的进展和严重的临床症状相关。
A novel KRIT1/CCM1 mutation accompanied by a NOTCH3 mutation in a Chinese family with multiple cerebral cavernous malformations.
Family cerebral cavernous malformations (FCCMs) are mainly inherited through the mutation of classical CCM genes, including CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10. FCCMs can cause severe clinical symptoms, including epileptic seizures, intracranial hemorrhage (ICH), or functional neurological deficits (FNDs). In this study, we reported a novel mutation in KRIT1 accompanied by a NOTCH3 mutation in a Chinese family. This family consists of 8 members, 4 of whom had been diagnosed with CCMs using cerebral MRI (T1WI, T2WI, SWI). The proband (II-2) and her daughter (III-4) had intracerebral hemorrhage and refractory epilepsy, respectively. Based on whole-exome sequencing (WES) data and bioinformatics analysis from 4 patients with multiple CCMs and 2 normal first-degree relatives, a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in intron 13 was considered a pathogenic gene in this family. Furthermore, based on 2 severe and 2 mild CCM patients, we found an SNV missense mutation, NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C), in NOTCH3. Finally, the KRIT1 and NOTCH3 mutations were validated in 8 members using Sanger sequencing. This study revealed a novel KRIT1 mutation, NG_012964.1 (NM_194456.1): c.1255-1G > T (splice-3), in a Chinese CCM family, which had not been reported previously. Moreover, the NOTCH3 mutation NG_009819.1 (NM_000435.2): c.1630C > T (p.R544C) might be a second hit and associated with the progression of CCM lesions and severe clinical symptoms.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


