{"title":"在阿尔法fold时代追求远程进化配对","authors":"Theodoros K. Karamanos","doi":"10.1002/bip.23530","DOIUrl":null,"url":null,"abstract":"<p>Coevolution between protein residues is normally interpreted as direct contact. However, the evolutionary record of a protein sequence contains rich information that may include long-range functional couplings, couplings that report on homo-oligomeric states or even conformational changes. Due to the complexity of the sequence space and the lack of structural information on various members of a protein family, it has been difficult to effectively mine the additional information encoded in a multiple sequence alignment (MSA). Here, taking advantage of the recent release of the AlphaFold (AF) database we attempt to identify coevolutionary couplings that cannot be explained simply by spatial proximity. We propose a simple computational method that performs direct coupling analysis on a MSA and searches for couplings that are not satisfied in any of the AF models of members of the identified protein family. Application of this method on 2012 protein families suggests that ~12% of the total identified coevolving residue pairs are spatially distant and more likely to be disordered than their contacting counterparts. We expect that this analysis will help improve the quality of coevolutionary distance restraints used for structure determination and will be useful in identifying potentially functional/allosteric cross-talk between distant residues.</p>","PeriodicalId":8866,"journal":{"name":"Biopolymers","volume":"114 3","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2023-02-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/bip.23530","citationCount":"2","resultStr":"{\"title\":\"Chasing long-range evolutionary couplings in the AlphaFold era\",\"authors\":\"Theodoros K. Karamanos\",\"doi\":\"10.1002/bip.23530\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Coevolution between protein residues is normally interpreted as direct contact. However, the evolutionary record of a protein sequence contains rich information that may include long-range functional couplings, couplings that report on homo-oligomeric states or even conformational changes. Due to the complexity of the sequence space and the lack of structural information on various members of a protein family, it has been difficult to effectively mine the additional information encoded in a multiple sequence alignment (MSA). Here, taking advantage of the recent release of the AlphaFold (AF) database we attempt to identify coevolutionary couplings that cannot be explained simply by spatial proximity. We propose a simple computational method that performs direct coupling analysis on a MSA and searches for couplings that are not satisfied in any of the AF models of members of the identified protein family. Application of this method on 2012 protein families suggests that ~12% of the total identified coevolving residue pairs are spatially distant and more likely to be disordered than their contacting counterparts. We expect that this analysis will help improve the quality of coevolutionary distance restraints used for structure determination and will be useful in identifying potentially functional/allosteric cross-talk between distant residues.</p>\",\"PeriodicalId\":8866,\"journal\":{\"name\":\"Biopolymers\",\"volume\":\"114 3\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-02-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/bip.23530\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biopolymers\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/bip.23530\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biopolymers","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/bip.23530","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Chasing long-range evolutionary couplings in the AlphaFold era
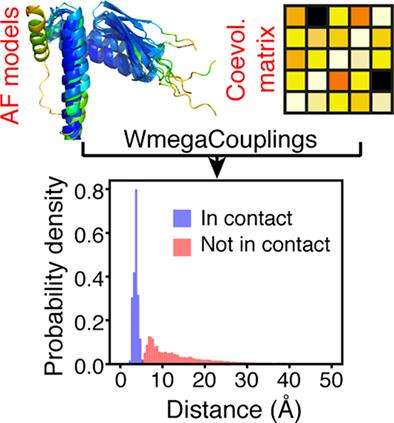
Coevolution between protein residues is normally interpreted as direct contact. However, the evolutionary record of a protein sequence contains rich information that may include long-range functional couplings, couplings that report on homo-oligomeric states or even conformational changes. Due to the complexity of the sequence space and the lack of structural information on various members of a protein family, it has been difficult to effectively mine the additional information encoded in a multiple sequence alignment (MSA). Here, taking advantage of the recent release of the AlphaFold (AF) database we attempt to identify coevolutionary couplings that cannot be explained simply by spatial proximity. We propose a simple computational method that performs direct coupling analysis on a MSA and searches for couplings that are not satisfied in any of the AF models of members of the identified protein family. Application of this method on 2012 protein families suggests that ~12% of the total identified coevolving residue pairs are spatially distant and more likely to be disordered than their contacting counterparts. We expect that this analysis will help improve the quality of coevolutionary distance restraints used for structure determination and will be useful in identifying potentially functional/allosteric cross-talk between distant residues.
期刊介绍:
Founded in 1963, Biopolymers publishes strictly peer-reviewed papers examining naturally occurring and synthetic biological macromolecules. By including experimental and theoretical studies on the fundamental behaviour as well as applications of biopolymers, the journal serves the interdisciplinary biochemical, biophysical, biomaterials and biomedical research communities.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


