Ida B K Suardana, Bayu K Mahardika, Made Pharmawati, Putu H Sudipa, Tri K Sari, Nyoman B Mahendra, Gusti N Mahardika
{"title":"SARS-CoV-2所有变体代表的全基因组比较,包括亚变体BA.2和GKA分支","authors":"Ida B K Suardana, Bayu K Mahardika, Made Pharmawati, Putu H Sudipa, Tri K Sari, Nyoman B Mahendra, Gusti N Mahardika","doi":"10.1155/2023/6476626","DOIUrl":null,"url":null,"abstract":"<p><p>Since its discovery at the end of 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly evolved into many variants, including the subvariant BA.2 and the GKA clade. Genomic clarification is needed for better management of the current pandemic as well as the possible reemergence of novel variants. The sequence of the reference genome Wuhan-Hu-1 and approximately 20 representatives of each variant were downloaded from GenBank and GISAID. Two representatives with no track of in-definitive nucleotides were selected. The sequences were aligned using muscle. The location of insertion/deletion (indel) in the genome was mapped following the open reading frame (ORF) of Wuhan-Hu-1. The phylogeny of the spike protein coding region was constructed using the maximum likelihood method. Amino acid substitutions in all ORFs were analyzed separately. There are two indel sites in ORF1AB, eight in spike, and one each in ORF3A, matrix (MA), nucleoprotein (NP), and the 3'-untranslated regions (3'UTR). Some indel sites and residues/substitutions are not unique, and some are variant-specific. The phylogeny shows that Omicron, Deltacron, and BA2 are clustered together and separated from other variants with 100% bootstrap support. In conclusion, whole-genome comparison of representatives of all variants revealed indel patterns that are specific to SARS-CoV-2 variants or subvariants. Polymorphic amino acid comparison across all coding regions also showed amino acid residues shared by specific groups of variants. Finally, the higher transmissibility of BA.2 might be due at least in part to the 48 nucleotide deletions in the 3'UTR, while the seem-to-be extinction of GKA clade is due to the lack of genetic advantages as a consequence of amino acid substitutions in various genes.</p>","PeriodicalId":7473,"journal":{"name":"Advances in Virology","volume":"2023 ","pages":"6476626"},"PeriodicalIF":1.4000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10019969/pdf/","citationCount":"1","resultStr":"{\"title\":\"Whole-Genome Comparison of Representatives of All Variants of SARS-CoV-2, Including Subvariant BA.2 and the GKA Clade.\",\"authors\":\"Ida B K Suardana, Bayu K Mahardika, Made Pharmawati, Putu H Sudipa, Tri K Sari, Nyoman B Mahendra, Gusti N Mahardika\",\"doi\":\"10.1155/2023/6476626\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Since its discovery at the end of 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly evolved into many variants, including the subvariant BA.2 and the GKA clade. Genomic clarification is needed for better management of the current pandemic as well as the possible reemergence of novel variants. The sequence of the reference genome Wuhan-Hu-1 and approximately 20 representatives of each variant were downloaded from GenBank and GISAID. Two representatives with no track of in-definitive nucleotides were selected. The sequences were aligned using muscle. The location of insertion/deletion (indel) in the genome was mapped following the open reading frame (ORF) of Wuhan-Hu-1. The phylogeny of the spike protein coding region was constructed using the maximum likelihood method. Amino acid substitutions in all ORFs were analyzed separately. There are two indel sites in ORF1AB, eight in spike, and one each in ORF3A, matrix (MA), nucleoprotein (NP), and the 3'-untranslated regions (3'UTR). Some indel sites and residues/substitutions are not unique, and some are variant-specific. The phylogeny shows that Omicron, Deltacron, and BA2 are clustered together and separated from other variants with 100% bootstrap support. In conclusion, whole-genome comparison of representatives of all variants revealed indel patterns that are specific to SARS-CoV-2 variants or subvariants. Polymorphic amino acid comparison across all coding regions also showed amino acid residues shared by specific groups of variants. Finally, the higher transmissibility of BA.2 might be due at least in part to the 48 nucleotide deletions in the 3'UTR, while the seem-to-be extinction of GKA clade is due to the lack of genetic advantages as a consequence of amino acid substitutions in various genes.</p>\",\"PeriodicalId\":7473,\"journal\":{\"name\":\"Advances in Virology\",\"volume\":\"2023 \",\"pages\":\"6476626\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10019969/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Advances in Virology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/6476626\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances in Virology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/6476626","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"VIROLOGY","Score":null,"Total":0}
Whole-Genome Comparison of Representatives of All Variants of SARS-CoV-2, Including Subvariant BA.2 and the GKA Clade.
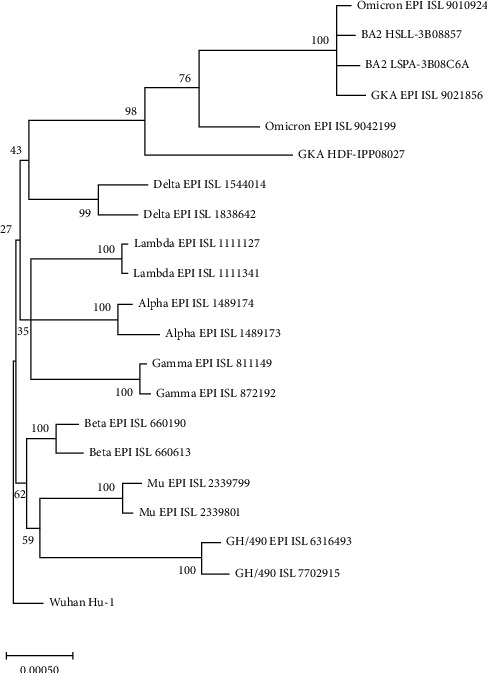
Since its discovery at the end of 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly evolved into many variants, including the subvariant BA.2 and the GKA clade. Genomic clarification is needed for better management of the current pandemic as well as the possible reemergence of novel variants. The sequence of the reference genome Wuhan-Hu-1 and approximately 20 representatives of each variant were downloaded from GenBank and GISAID. Two representatives with no track of in-definitive nucleotides were selected. The sequences were aligned using muscle. The location of insertion/deletion (indel) in the genome was mapped following the open reading frame (ORF) of Wuhan-Hu-1. The phylogeny of the spike protein coding region was constructed using the maximum likelihood method. Amino acid substitutions in all ORFs were analyzed separately. There are two indel sites in ORF1AB, eight in spike, and one each in ORF3A, matrix (MA), nucleoprotein (NP), and the 3'-untranslated regions (3'UTR). Some indel sites and residues/substitutions are not unique, and some are variant-specific. The phylogeny shows that Omicron, Deltacron, and BA2 are clustered together and separated from other variants with 100% bootstrap support. In conclusion, whole-genome comparison of representatives of all variants revealed indel patterns that are specific to SARS-CoV-2 variants or subvariants. Polymorphic amino acid comparison across all coding regions also showed amino acid residues shared by specific groups of variants. Finally, the higher transmissibility of BA.2 might be due at least in part to the 48 nucleotide deletions in the 3'UTR, while the seem-to-be extinction of GKA clade is due to the lack of genetic advantages as a consequence of amino acid substitutions in various genes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


