David Henke, Felipe-Andrés Piedra, Vasanthi Avadhanula, Harsha Doddapaneni, Donna M Muzny, Vipin K Menon, Kristi L Hoffman, Matthew C Ross, Sara J Javornik Cregeen, Ginger Metcalf, Richard A Gibbs, Joseph F Petrosino, Pedro A Piedra
{"title":"通过短读数高通量测序研究 RSV 的宿主内遗传变异。","authors":"David Henke, Felipe-Andrés Piedra, Vasanthi Avadhanula, Harsha Doddapaneni, Donna M Muzny, Vipin K Menon, Kristi L Hoffman, Matthew C Ross, Sara J Javornik Cregeen, Ginger Metcalf, Richard A Gibbs, Joseph F Petrosino, Pedro A Piedra","doi":"10.1101/2023.05.17.541198","DOIUrl":null,"url":null,"abstract":"<p><p>Every viral infection entails an evolving population of viral genomes. High-throughput sequencing technologies can be used to characterize such populations, but to date there are few published examples of such work. In addition, mixed sequencing data are sometimes used to infer properties of infecting genomes without discriminating between genome-derived reads and reads from the much more abundant, in the case of a typical active viral infection, transcripts. Here we apply capture probe-based short read high-throughput sequencing to nasal wash samples taken from a previously described group of adult hematopoietic cell transplant (HCT) recipients naturally infected with respiratory syncytial virus (RSV). We separately analyzed reads from genomes and transcripts for the levels and distribution of genetic variation by calculating per position Shannon entropies. Our analysis reveals a low level of genetic variation within the RSV infections analyzed here, but with interesting differences between genomes and transcripts in 1) average per sample Shannon entropies; 2) the genomic distribution of variation 'hotspots'; and 3) the genomic distribution of hotspots encoding alternative amino acids. In all, our results suggest the importance of separately analyzing reads from genomes and transcripts when interpreting high-throughput sequencing data for insight into intra-host viral genome replication, expression, and evolution.</p>","PeriodicalId":43223,"journal":{"name":"Revista CS en Ciencias Sociales","volume":"1 1","pages":""},"PeriodicalIF":0.3000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11398394/pdf/","citationCount":"0","resultStr":"{\"title\":\"Examining intra-host genetic variation of RSV by short read high-throughput sequencing.\",\"authors\":\"David Henke, Felipe-Andrés Piedra, Vasanthi Avadhanula, Harsha Doddapaneni, Donna M Muzny, Vipin K Menon, Kristi L Hoffman, Matthew C Ross, Sara J Javornik Cregeen, Ginger Metcalf, Richard A Gibbs, Joseph F Petrosino, Pedro A Piedra\",\"doi\":\"10.1101/2023.05.17.541198\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Every viral infection entails an evolving population of viral genomes. High-throughput sequencing technologies can be used to characterize such populations, but to date there are few published examples of such work. In addition, mixed sequencing data are sometimes used to infer properties of infecting genomes without discriminating between genome-derived reads and reads from the much more abundant, in the case of a typical active viral infection, transcripts. Here we apply capture probe-based short read high-throughput sequencing to nasal wash samples taken from a previously described group of adult hematopoietic cell transplant (HCT) recipients naturally infected with respiratory syncytial virus (RSV). We separately analyzed reads from genomes and transcripts for the levels and distribution of genetic variation by calculating per position Shannon entropies. Our analysis reveals a low level of genetic variation within the RSV infections analyzed here, but with interesting differences between genomes and transcripts in 1) average per sample Shannon entropies; 2) the genomic distribution of variation 'hotspots'; and 3) the genomic distribution of hotspots encoding alternative amino acids. In all, our results suggest the importance of separately analyzing reads from genomes and transcripts when interpreting high-throughput sequencing data for insight into intra-host viral genome replication, expression, and evolution.</p>\",\"PeriodicalId\":43223,\"journal\":{\"name\":\"Revista CS en Ciencias Sociales\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":0.3000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11398394/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Revista CS en Ciencias Sociales\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.05.17.541198\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"SOCIAL ISSUES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista CS en Ciencias Sociales","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.05.17.541198","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"SOCIAL ISSUES","Score":null,"Total":0}
Examining intra-host genetic variation of RSV by short read high-throughput sequencing.
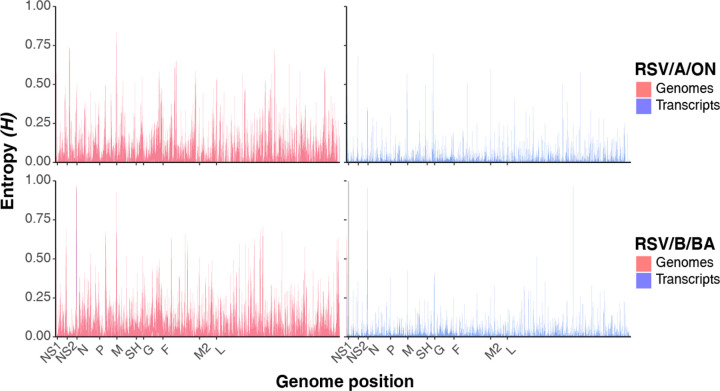
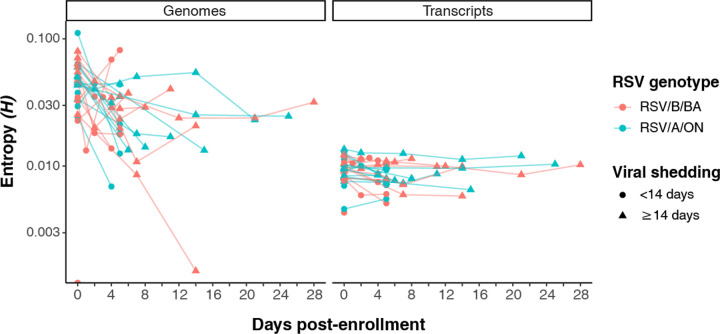
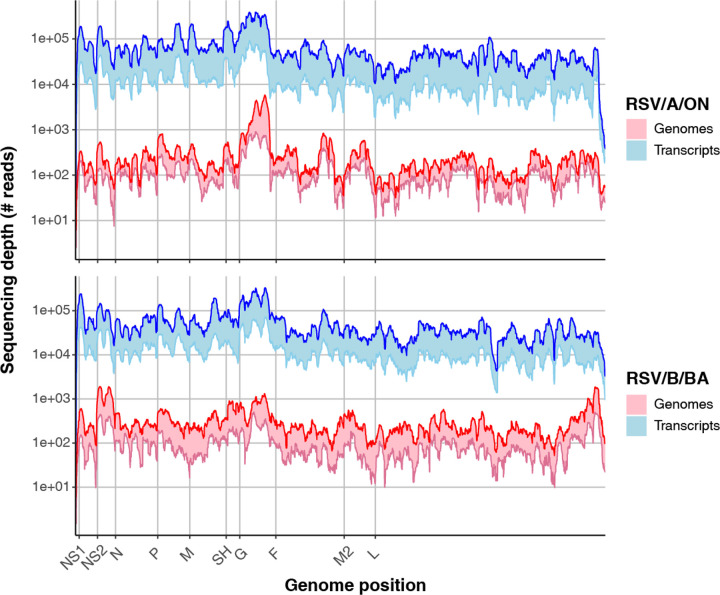
Every viral infection entails an evolving population of viral genomes. High-throughput sequencing technologies can be used to characterize such populations, but to date there are few published examples of such work. In addition, mixed sequencing data are sometimes used to infer properties of infecting genomes without discriminating between genome-derived reads and reads from the much more abundant, in the case of a typical active viral infection, transcripts. Here we apply capture probe-based short read high-throughput sequencing to nasal wash samples taken from a previously described group of adult hematopoietic cell transplant (HCT) recipients naturally infected with respiratory syncytial virus (RSV). We separately analyzed reads from genomes and transcripts for the levels and distribution of genetic variation by calculating per position Shannon entropies. Our analysis reveals a low level of genetic variation within the RSV infections analyzed here, but with interesting differences between genomes and transcripts in 1) average per sample Shannon entropies; 2) the genomic distribution of variation 'hotspots'; and 3) the genomic distribution of hotspots encoding alternative amino acids. In all, our results suggest the importance of separately analyzing reads from genomes and transcripts when interpreting high-throughput sequencing data for insight into intra-host viral genome replication, expression, and evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


