Sebastian Finke, Anja Stammler, Jan Oldengott, Stephan Walleck and Thorsten Glaser
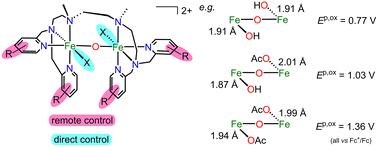
{"title":"μ-氧二铁配合物电子结构和氧化还原电位的直接和远程控制","authors":"Sebastian Finke, Anja Stammler, Jan Oldengott, Stephan Walleck and Thorsten Glaser","doi":"10.1039/D3DT02734A","DOIUrl":null,"url":null,"abstract":"<p >Non-heme diiron enzymes activate O<small><sub>2</sub></small> for the oxidation of substrates in the form of peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> or high-valent Fe<small><sup>IV</sup></small><small><sub>2</sub></small> intermediates. We have developed a dinucleating bis(tetradentate) ligand system that stabilizes peroxo and hydroperoxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> complexes with terminal 6-methylpyridine donors, while the peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> intermediate is reactive with terminal pyridine donors presumably <em>via</em> conversion to a fluent high-valent Fe<small><sup>IV</sup></small><small><sub>2</sub></small> intermediate. We present here a derivative with electron-donating methoxy substituents at the pyridine donors and its diferric complexes with an {Fe<small><sup>III</sup></small>X(μ-O)Fe<small><sup>III</sup></small>X} (X<small><sup>−</sup></small> = Cl<small><sup>−</sup></small>, OAc<small><sup>−</sup></small>, and OH<small><sup>−</sup></small>) or an {Fe<small><sup>III</sup></small>(μ-O)(μ-OAc)Fe<small><sup>III</sup></small>} core. The complex-induced oxidation of EtOH with H<small><sub>2</sub></small>O<small><sub>2</sub></small> provides μ-OAc<small><sup>−</sup></small>, and in acetone, the complex with mixed OH<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> exogenous donors is obtained. Both reactivities indicate a reactive fluent peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> intermediate. The coupling constant <em>J</em> and the LMCT transitions are insensitive to the nature of the directly bound ligands X<small><sup>−</sup></small> and reflect mainly the electronic structure of the central {Fe<small><sup>III</sup></small>(μ-O)Fe<small><sup>III</sup></small>} core, while Mössbauer spectroscopy and d–d transitions probe the local Fe<small><sup>III</sup></small> sites. The remote methoxy substituents decrease the potential for the oxidation to Fe<small><sup>IV</sup></small> by ∼100 mV, while directly bound OH<small><sup>−</sup></small> in {Fe<small><sup>III</sup></small>(OH)(μ-O)Fe<small><sup>III</sup></small>(OH)} with a short 1.91 Å Fe<small><sup>III</sup></small>–O<small><sup>OH</sup></small> bond decreases the potential by 590 mV compared to {Fe<small><sup>III</sup></small>(OAc)(μ-O)Fe<small><sup>III</sup></small>(OAc)} with a 2.01 Å Fe<small><sup>III</sup></small>–O<small><sup>OAc</sup></small> bond. Interestingly, this Fe<small><sup>III</sup></small>–OH bond is even shorter (1.87 Å) in the mixed OH<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> complex but the potential is the mean value of the potentials of the OH<small><sup>−</sup></small>/OH<small><sup>−</sup></small> and OAc<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> complexes, thus reflecting the electron density of the central {Fe<small><sup>III</sup></small>(μ-O)Fe<small><sup>III</sup></small>} core and not of the local Fe<small><sup>III</sup></small>–OH unit.</p>","PeriodicalId":71,"journal":{"name":"Dalton Transactions","volume":" 46","pages":" 17548-17561"},"PeriodicalIF":3.5000,"publicationDate":"2023-11-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Direct and remote control of electronic structures and redox potentials in μ-oxo diferric complexes†\",\"authors\":\"Sebastian Finke, Anja Stammler, Jan Oldengott, Stephan Walleck and Thorsten Glaser\",\"doi\":\"10.1039/D3DT02734A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Non-heme diiron enzymes activate O<small><sub>2</sub></small> for the oxidation of substrates in the form of peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> or high-valent Fe<small><sup>IV</sup></small><small><sub>2</sub></small> intermediates. We have developed a dinucleating bis(tetradentate) ligand system that stabilizes peroxo and hydroperoxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> complexes with terminal 6-methylpyridine donors, while the peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> intermediate is reactive with terminal pyridine donors presumably <em>via</em> conversion to a fluent high-valent Fe<small><sup>IV</sup></small><small><sub>2</sub></small> intermediate. We present here a derivative with electron-donating methoxy substituents at the pyridine donors and its diferric complexes with an {Fe<small><sup>III</sup></small>X(μ-O)Fe<small><sup>III</sup></small>X} (X<small><sup>−</sup></small> = Cl<small><sup>−</sup></small>, OAc<small><sup>−</sup></small>, and OH<small><sup>−</sup></small>) or an {Fe<small><sup>III</sup></small>(μ-O)(μ-OAc)Fe<small><sup>III</sup></small>} core. The complex-induced oxidation of EtOH with H<small><sub>2</sub></small>O<small><sub>2</sub></small> provides μ-OAc<small><sup>−</sup></small>, and in acetone, the complex with mixed OH<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> exogenous donors is obtained. Both reactivities indicate a reactive fluent peroxo Fe<small><sup>III</sup></small><small><sub>2</sub></small> intermediate. The coupling constant <em>J</em> and the LMCT transitions are insensitive to the nature of the directly bound ligands X<small><sup>−</sup></small> and reflect mainly the electronic structure of the central {Fe<small><sup>III</sup></small>(μ-O)Fe<small><sup>III</sup></small>} core, while Mössbauer spectroscopy and d–d transitions probe the local Fe<small><sup>III</sup></small> sites. The remote methoxy substituents decrease the potential for the oxidation to Fe<small><sup>IV</sup></small> by ∼100 mV, while directly bound OH<small><sup>−</sup></small> in {Fe<small><sup>III</sup></small>(OH)(μ-O)Fe<small><sup>III</sup></small>(OH)} with a short 1.91 Å Fe<small><sup>III</sup></small>–O<small><sup>OH</sup></small> bond decreases the potential by 590 mV compared to {Fe<small><sup>III</sup></small>(OAc)(μ-O)Fe<small><sup>III</sup></small>(OAc)} with a 2.01 Å Fe<small><sup>III</sup></small>–O<small><sup>OAc</sup></small> bond. Interestingly, this Fe<small><sup>III</sup></small>–OH bond is even shorter (1.87 Å) in the mixed OH<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> complex but the potential is the mean value of the potentials of the OH<small><sup>−</sup></small>/OH<small><sup>−</sup></small> and OAc<small><sup>−</sup></small>/OAc<small><sup>−</sup></small> complexes, thus reflecting the electron density of the central {Fe<small><sup>III</sup></small>(μ-O)Fe<small><sup>III</sup></small>} core and not of the local Fe<small><sup>III</sup></small>–OH unit.</p>\",\"PeriodicalId\":71,\"journal\":{\"name\":\"Dalton Transactions\",\"volume\":\" 46\",\"pages\":\" 17548-17561\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2023-11-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Dalton Transactions\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/dt/d3dt02734a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Dalton Transactions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/dt/d3dt02734a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
摘要
非血红素二铁酶激活O2以过氧FeIII2或高价FeIV2中间体的形式氧化底物。我们已经开发了一种双核双(四齿)配体系统,该系统稳定具有末端6-甲基吡啶供体的过氧和氢过氧FeIII2复合物,而过氧FeIII3中间体可能通过转化为流畅的高价FeIV2中间体与末端吡啶供体反应。我们在此提出了在吡啶供体上具有给电子甲氧基取代基的衍生物,以及它与{FeIIIX(μ-O)FeIIIX}(X=Cl-、OAc-和OH-)或{FeIII(μ-O)(μ-OAc)FeIII}核的二铁络合物。配合物诱导EtOH与H2O2的氧化提供了μ-OAc-,并在丙酮中获得了具有混合OH-/OAc-外源供体的配合物。两种反应性都表明存在反应性流动的过氧FeIII2中间体。耦合常数J和LMCT跃迁对直接结合配体X-的性质不敏感,主要反映中心{FeIII(μ-O)FeIII}核的电子结构,而穆斯堡尔谱和d-d跃迁探测局部FeIII位点。远端甲氧基取代基将氧化成FeIV的电位降低约100 mV,而与具有2.01ÅFeIII OOAc键的{FeIII(OAc)(μ-O)FeIII(OAc)}相比,具有短1.91ÅFe III OOH键的{FeIII(OH)(μ.O)FeIII(OH)}中直接结合的OH-将电位降低590 mV。有趣的是,在混合OH-/OAc-复合物中,这种FeIII-OH键甚至更短(1.87Å),但电势是OH-/OH-和OAc-/OAc-复合物电势的平均值,因此反映了中心{FeIII(μ-O)FeIII}核的电子密度,而不是局部FeIII-OH-单元的电子密度。
Direct and remote control of electronic structures and redox potentials in μ-oxo diferric complexes†
Non-heme diiron enzymes activate O2 for the oxidation of substrates in the form of peroxo FeIII2 or high-valent FeIV2 intermediates. We have developed a dinucleating bis(tetradentate) ligand system that stabilizes peroxo and hydroperoxo FeIII2 complexes with terminal 6-methylpyridine donors, while the peroxo FeIII2 intermediate is reactive with terminal pyridine donors presumably via conversion to a fluent high-valent FeIV2 intermediate. We present here a derivative with electron-donating methoxy substituents at the pyridine donors and its diferric complexes with an {FeIIIX(μ-O)FeIIIX} (X− = Cl−, OAc−, and OH−) or an {FeIII(μ-O)(μ-OAc)FeIII} core. The complex-induced oxidation of EtOH with H2O2 provides μ-OAc−, and in acetone, the complex with mixed OH−/OAc− exogenous donors is obtained. Both reactivities indicate a reactive fluent peroxo FeIII2 intermediate. The coupling constant J and the LMCT transitions are insensitive to the nature of the directly bound ligands X− and reflect mainly the electronic structure of the central {FeIII(μ-O)FeIII} core, while Mössbauer spectroscopy and d–d transitions probe the local FeIII sites. The remote methoxy substituents decrease the potential for the oxidation to FeIV by ∼100 mV, while directly bound OH− in {FeIII(OH)(μ-O)FeIII(OH)} with a short 1.91 Å FeIII–OOH bond decreases the potential by 590 mV compared to {FeIII(OAc)(μ-O)FeIII(OAc)} with a 2.01 Å FeIII–OOAc bond. Interestingly, this FeIII–OH bond is even shorter (1.87 Å) in the mixed OH−/OAc− complex but the potential is the mean value of the potentials of the OH−/OH− and OAc−/OAc− complexes, thus reflecting the electron density of the central {FeIII(μ-O)FeIII} core and not of the local FeIII–OH unit.
期刊介绍:
Dalton Transactions is a journal for all areas of inorganic chemistry, which encompasses the organometallic, bioinorganic and materials chemistry of the elements, with applications including synthesis, catalysis, energy conversion/storage, electrical devices and medicine. Dalton Transactions welcomes high-quality, original submissions in all of these areas and more, where the advancement of knowledge in inorganic chemistry is significant.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


