Ildikó Tar, Márta Szegedi, Ewa Krasuska-Sławińska, Edyta Heropolitańska-Pliszka, Ewa A Bernatowska, Elif Öncü, Sevgi Keles, Sukru N Guner, Ismail Reisli, Nevena Gesheva, Elissaveta Naumova, Lydie Izakovicova-Holla, Jiri Litzman, Igor Savchak, Larysa Kostyuchenko, Melinda Erdõs
{"title":"常染色体显性高IgE综合征患者口腔颌面部异常。","authors":"Ildikó Tar, Márta Szegedi, Ewa Krasuska-Sławińska, Edyta Heropolitańska-Pliszka, Ewa A Bernatowska, Elif Öncü, Sevgi Keles, Sukru N Guner, Ismail Reisli, Nevena Gesheva, Elissaveta Naumova, Lydie Izakovicova-Holla, Jiri Litzman, Igor Savchak, Larysa Kostyuchenko, Melinda Erdõs","doi":"10.5114/ceji.2023.130874","DOIUrl":null,"url":null,"abstract":"<p><p>Autosomal dominant hyper-IgE syndrome (AD-HIES) is an inborn error of immunity (IEI) caused by a dominant-negative mutation in the signal transducer and activator of transcription 3 (STAT 3). This disease is characterized by chronic eczematoid dermatitis, recurrent staphylococcal skin abscesses, pneumonia, pneumatoceles, and extremely high serum IgE levels. Loss-of-function STAT3 mutations may also result in distinct non-immunologic features such as dental, facial, skeletal, and vascular abnormalities, central nervous system malformations and an increased risk for bone fractures. Prophylactic treatment of Candida infections and prophylactic antimicrobial therapy for staphylococcal skin infections and sinopulmonary infections are essential. An awareness of the oral and maxillofacial features of HIES may facilitate early diagnosis with genetic counselling and may improve future patient care. This study describes oral, dental, and maxillofacial manifestations in 14 patients with genetically defined AD-HIES. We also review the literature and propose recommendations for the complex care of patients with this rare primary immunodeficiency.</p>","PeriodicalId":9694,"journal":{"name":"Central European Journal of Immunology","volume":"1 1","pages":"228-236"},"PeriodicalIF":1.6000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10604639/pdf/","citationCount":"0","resultStr":"{\"title\":\"Intraoral and maxillofacial abnormalities in patients with autosomal dominant hyper-IgE syndrome.\",\"authors\":\"Ildikó Tar, Márta Szegedi, Ewa Krasuska-Sławińska, Edyta Heropolitańska-Pliszka, Ewa A Bernatowska, Elif Öncü, Sevgi Keles, Sukru N Guner, Ismail Reisli, Nevena Gesheva, Elissaveta Naumova, Lydie Izakovicova-Holla, Jiri Litzman, Igor Savchak, Larysa Kostyuchenko, Melinda Erdõs\",\"doi\":\"10.5114/ceji.2023.130874\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Autosomal dominant hyper-IgE syndrome (AD-HIES) is an inborn error of immunity (IEI) caused by a dominant-negative mutation in the signal transducer and activator of transcription 3 (STAT 3). This disease is characterized by chronic eczematoid dermatitis, recurrent staphylococcal skin abscesses, pneumonia, pneumatoceles, and extremely high serum IgE levels. Loss-of-function STAT3 mutations may also result in distinct non-immunologic features such as dental, facial, skeletal, and vascular abnormalities, central nervous system malformations and an increased risk for bone fractures. Prophylactic treatment of Candida infections and prophylactic antimicrobial therapy for staphylococcal skin infections and sinopulmonary infections are essential. An awareness of the oral and maxillofacial features of HIES may facilitate early diagnosis with genetic counselling and may improve future patient care. This study describes oral, dental, and maxillofacial manifestations in 14 patients with genetically defined AD-HIES. We also review the literature and propose recommendations for the complex care of patients with this rare primary immunodeficiency.</p>\",\"PeriodicalId\":9694,\"journal\":{\"name\":\"Central European Journal of Immunology\",\"volume\":\"1 1\",\"pages\":\"228-236\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10604639/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Central European Journal of Immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.5114/ceji.2023.130874\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Central European Journal of Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.5114/ceji.2023.130874","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/5 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Intraoral and maxillofacial abnormalities in patients with autosomal dominant hyper-IgE syndrome.
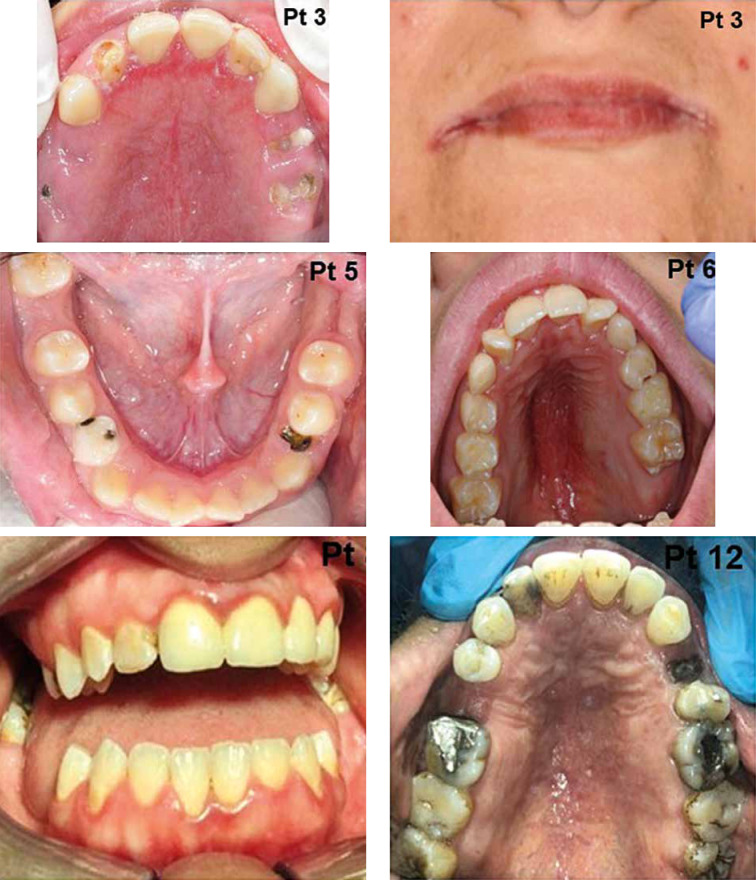
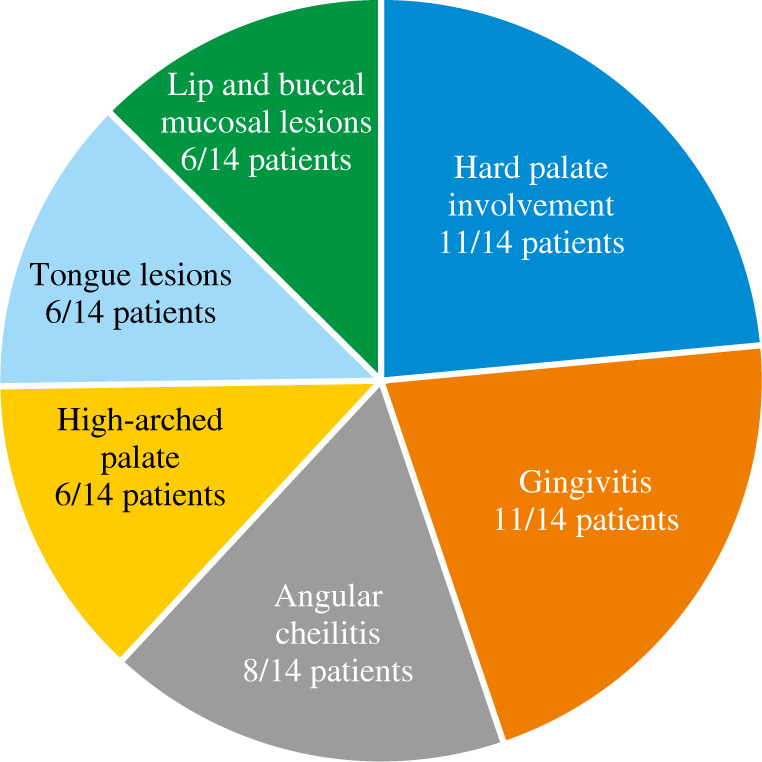
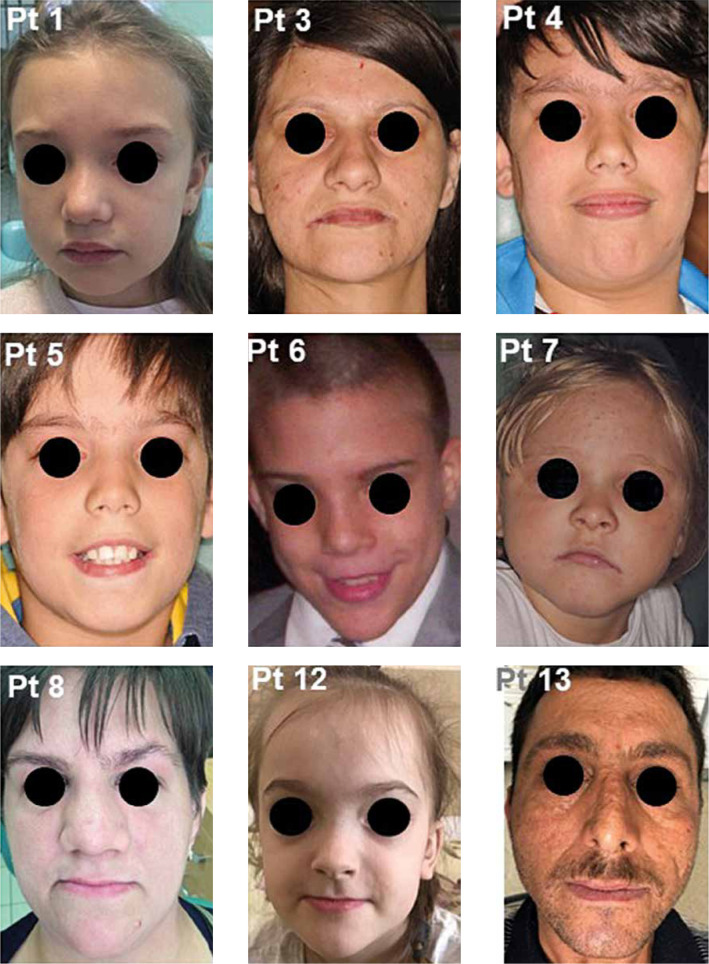
Autosomal dominant hyper-IgE syndrome (AD-HIES) is an inborn error of immunity (IEI) caused by a dominant-negative mutation in the signal transducer and activator of transcription 3 (STAT 3). This disease is characterized by chronic eczematoid dermatitis, recurrent staphylococcal skin abscesses, pneumonia, pneumatoceles, and extremely high serum IgE levels. Loss-of-function STAT3 mutations may also result in distinct non-immunologic features such as dental, facial, skeletal, and vascular abnormalities, central nervous system malformations and an increased risk for bone fractures. Prophylactic treatment of Candida infections and prophylactic antimicrobial therapy for staphylococcal skin infections and sinopulmonary infections are essential. An awareness of the oral and maxillofacial features of HIES may facilitate early diagnosis with genetic counselling and may improve future patient care. This study describes oral, dental, and maxillofacial manifestations in 14 patients with genetically defined AD-HIES. We also review the literature and propose recommendations for the complex care of patients with this rare primary immunodeficiency.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


