Vladimir Perovic, Sanja Glisic, Milena Veljkovic, Slobodan Paessler, Veljko Veljkovic
{"title":"一种新的基于熵的系统发育算法:对严重急性呼吸系统综合征冠状病毒2型变异株进行分类的新方法。","authors":"Vladimir Perovic, Sanja Glisic, Milena Veljkovic, Slobodan Paessler, Veljko Veljkovic","doi":"10.3390/e25101463","DOIUrl":null,"url":null,"abstract":"<p><p>The SARS-CoV-2 virus, the causative agent of COVID-19, is known for its genetic diversity. Virus variants of concern (VOCs) as well as variants of interest (VOIs) are classified by the World Health Organization (WHO) according to their potential risk to global health. This study seeks to enhance the identification and classification of such variants by developing a novel bioinformatics criterion centered on the virus's spike protein (SP1), a key player in host cell entry, immune response, and a mutational hotspot. To achieve this, we pioneered a unique phylogenetic algorithm which calculates EIIP-entropy as a distance measure based on the distribution of the electron-ion interaction potential (EIIP) of amino acids in SP1. This method offers a comprehensive, scalable, and rapid approach to analyze large genomic data sets and predict the impact of specific mutations. This innovative approach provides a robust tool for classifying emergent SARS-CoV-2 variants into potential VOCs or VOIs. It could significantly augment surveillance efforts and understanding of variant characteristics, while also offering potential applicability to the analysis and classification of other emerging viral pathogens and enhancing global readiness against emerging and re-emerging viral pathogens.</p>","PeriodicalId":11694,"journal":{"name":"Entropy","volume":"25 10","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2023-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10606860/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel Entropy-Based Phylogenetic Algorithm: A New Approach for Classifying SARS-CoV-2 Variants.\",\"authors\":\"Vladimir Perovic, Sanja Glisic, Milena Veljkovic, Slobodan Paessler, Veljko Veljkovic\",\"doi\":\"10.3390/e25101463\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The SARS-CoV-2 virus, the causative agent of COVID-19, is known for its genetic diversity. Virus variants of concern (VOCs) as well as variants of interest (VOIs) are classified by the World Health Organization (WHO) according to their potential risk to global health. This study seeks to enhance the identification and classification of such variants by developing a novel bioinformatics criterion centered on the virus's spike protein (SP1), a key player in host cell entry, immune response, and a mutational hotspot. To achieve this, we pioneered a unique phylogenetic algorithm which calculates EIIP-entropy as a distance measure based on the distribution of the electron-ion interaction potential (EIIP) of amino acids in SP1. This method offers a comprehensive, scalable, and rapid approach to analyze large genomic data sets and predict the impact of specific mutations. This innovative approach provides a robust tool for classifying emergent SARS-CoV-2 variants into potential VOCs or VOIs. It could significantly augment surveillance efforts and understanding of variant characteristics, while also offering potential applicability to the analysis and classification of other emerging viral pathogens and enhancing global readiness against emerging and re-emerging viral pathogens.</p>\",\"PeriodicalId\":11694,\"journal\":{\"name\":\"Entropy\",\"volume\":\"25 10\",\"pages\":\"\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2023-10-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10606860/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Entropy\",\"FirstCategoryId\":\"101\",\"ListUrlMain\":\"https://doi.org/10.3390/e25101463\",\"RegionNum\":3,\"RegionCategory\":\"物理与天体物理\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PHYSICS, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Entropy","FirstCategoryId":"101","ListUrlMain":"https://doi.org/10.3390/e25101463","RegionNum":3,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PHYSICS, MULTIDISCIPLINARY","Score":null,"Total":0}
Novel Entropy-Based Phylogenetic Algorithm: A New Approach for Classifying SARS-CoV-2 Variants.
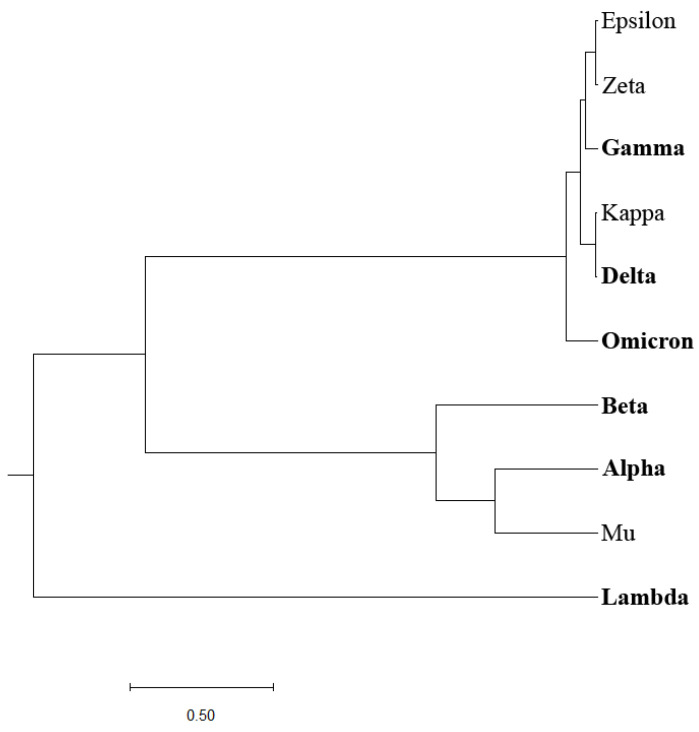
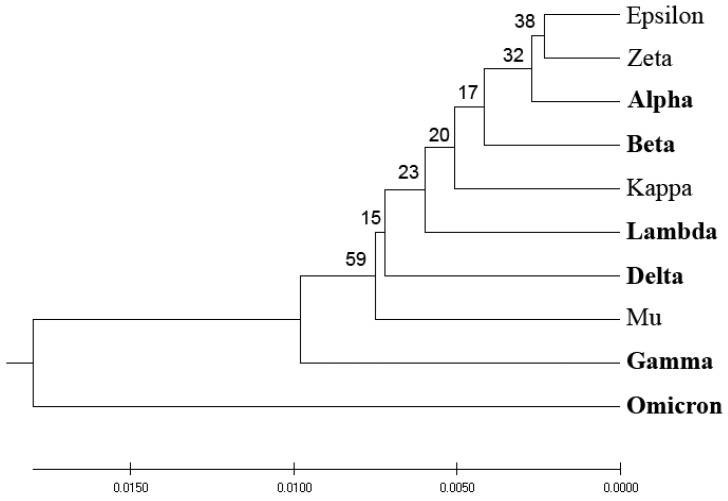
The SARS-CoV-2 virus, the causative agent of COVID-19, is known for its genetic diversity. Virus variants of concern (VOCs) as well as variants of interest (VOIs) are classified by the World Health Organization (WHO) according to their potential risk to global health. This study seeks to enhance the identification and classification of such variants by developing a novel bioinformatics criterion centered on the virus's spike protein (SP1), a key player in host cell entry, immune response, and a mutational hotspot. To achieve this, we pioneered a unique phylogenetic algorithm which calculates EIIP-entropy as a distance measure based on the distribution of the electron-ion interaction potential (EIIP) of amino acids in SP1. This method offers a comprehensive, scalable, and rapid approach to analyze large genomic data sets and predict the impact of specific mutations. This innovative approach provides a robust tool for classifying emergent SARS-CoV-2 variants into potential VOCs or VOIs. It could significantly augment surveillance efforts and understanding of variant characteristics, while also offering potential applicability to the analysis and classification of other emerging viral pathogens and enhancing global readiness against emerging and re-emerging viral pathogens.
期刊介绍:
Entropy (ISSN 1099-4300), an international and interdisciplinary journal of entropy and information studies, publishes reviews, regular research papers and short notes. Our aim is to encourage scientists to publish as much as possible their theoretical and experimental details. There is no restriction on the length of the papers. If there are computation and the experiment, the details must be provided so that the results can be reproduced.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


