使用GROMACS软件进行核磁共振细化和肽折叠
IF 1.3
3区 生物学
Q3 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 8
摘要
核磁共振波谱通常用于研究蛋白质和核酸的三维结构和动力学。结构的确定通常是通过在经典能量函数中加入基于核磁共振数据的约束并进行约束分子模拟来完成的。在这里,我们报告了一个从NMR- star文件中提取NMR约束并将其导出到GROMACS软件的脚本的实现。有了这个软件包,就可以对距离约束、二面体约束和方向约束进行建模。通过执行有约束和没有约束的模拟来验证脚本的输出,包括从头开始细化一个肽。本文章由计算机程序翻译,如有差异,请以英文原文为准。
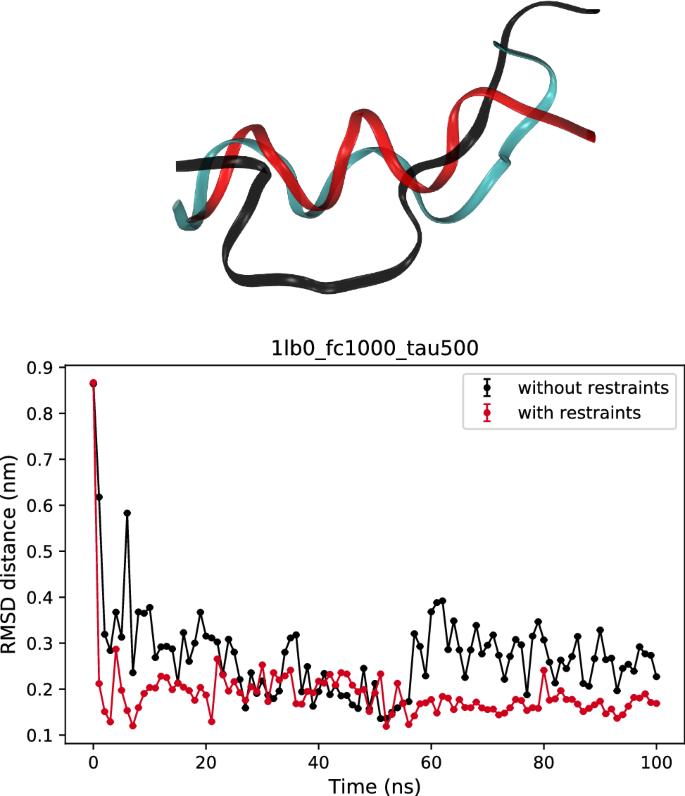
NMR refinement and peptide folding using the GROMACS software
Nuclear magnetic resonance spectroscopy is used routinely for studying the three-dimensional structures and dynamics of proteins and nucleic acids. Structure determination is usually done by adding restraints based upon NMR data to a classical energy function and performing restrained molecular simulations. Here we report on the implementation of a script to extract NMR restraints from a NMR-STAR file and export it to the GROMACS software. With this package it is possible to model distance restraints, dihedral restraints and orientation restraints. The output from the script is validated by performing simulations with and without restraints, including the ab initio refinement of one peptide.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Biomolecular NMR
生物-光谱学
CiteScore
6.00
自引率
3.70%
发文量
19
审稿时长
6-12 weeks
期刊介绍:
The Journal of Biomolecular NMR provides a forum for publishing research on technical developments and innovative applications of nuclear magnetic resonance spectroscopy for the study of structure and dynamic properties of biopolymers in solution, liquid crystals, solids and mixed environments, e.g., attached to membranes. This may include:
Three-dimensional structure determination of biological macromolecules (polypeptides/proteins, DNA, RNA, oligosaccharides) by NMR.
New NMR techniques for studies of biological macromolecules.
Novel approaches to computer-aided automated analysis of multidimensional NMR spectra.
Computational methods for the structural interpretation of NMR data, including structure refinement.
Comparisons of structures determined by NMR with those obtained by other methods, e.g. by diffraction techniques with protein single crystals.
New techniques of sample preparation for NMR experiments (biosynthetic and chemical methods for isotope labeling, preparation of nutrients for biosynthetic isotope labeling, etc.). An NMR characterization of the products must be included.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


