Iliana Michailidou , Jeroen Vreijling , Matthijs Rumpf , Maarten Loos , Bastijn Koopmans , Nina Vlek , Nina Straat , Cedrick Agaser , Thomas B. Kuipers , Hailiang Mei , Frank Baas , Kees Fluiter
{"title":"在PMP22过表达的CMT1A小鼠模型中,终末补体系统的全身性抑制可减少神经炎症,但不能改善运动功能","authors":"Iliana Michailidou , Jeroen Vreijling , Matthijs Rumpf , Maarten Loos , Bastijn Koopmans , Nina Vlek , Nina Straat , Cedrick Agaser , Thomas B. Kuipers , Hailiang Mei , Frank Baas , Kees Fluiter","doi":"10.1016/j.crneur.2023.100077","DOIUrl":null,"url":null,"abstract":"<div><p>Charcot-Marie-Tooth disease type 1A (CMT1A) is the most prevalent hereditary demyelinating neuropathy. This autosomal, dominantly inherited disease is caused by a duplication on chromosome 17p which includes the peripheral myelin protein 22 (PMP22) gene. There is clinical evidence that the disability in CMT1A is to a large extend due to axonal damage rather than demyelination. Over-expression of <em>PMP22</em> is recently thought to impede cholesterol trafficking causing a total shutdown of local cholesterol and lipid synthesis in the Schwann cells, thus disturbing their ability to remyelinate. But there is a large variety in disease burden between CMT1A patients with the same genetic defect, indicating the presence of modifying factors that affect disease severity. One of these potential factors is the immune system. Several reports have described patients with co-occurrence of CMT1A with chronic inflammatory demyelinating disease or Guillain-Barré syndrome. We have previously shown in multiple animal models that the innate immune system and specifically the terminal complement system is a driver of inflammatory demyelination. To test the contribution of the terminal complement system to neuroinflammation and disease progression in CMT1A, we inhibited systemic complement C6 in two transgenic mouse models for CMT1A, the C3-<em>PMP22</em> and C3-<em>PMP22</em> c-JunP0Cre models. Both models over-express human <em>PMP22</em>, and one (C3-<em>PMP22</em> c-JunP0Cre) also has a Schwann cell-specific knockout of c-Jun, a crucial regulator of myelination controlling autophagy. We found that systemic inhibition of C6 using antisense oligonucleotides affects the neuroinflammation, Rho GTPase and ERK/MAPK signalling pathways in the CMT1A mouse models. The cholesterol synthesis pathway remained unaffected. Analysis of motor function during treatment with C6 antisense oligonucleotides did not reveal any significant improvement in the CMT1A mouse models. This study shows that the contribution of the terminal complement system to progressive loss of motor function in the CMT1A mouse models tested is limited.</p></div>","PeriodicalId":72752,"journal":{"name":"Current research in neurobiology","volume":"4 ","pages":"Article 100077"},"PeriodicalIF":0.0000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The systemic inhibition of the terminal complement system reduces neuroinflammation but does not improve motor function in mouse models of CMT1A with overexpressed PMP22\",\"authors\":\"Iliana Michailidou , Jeroen Vreijling , Matthijs Rumpf , Maarten Loos , Bastijn Koopmans , Nina Vlek , Nina Straat , Cedrick Agaser , Thomas B. Kuipers , Hailiang Mei , Frank Baas , Kees Fluiter\",\"doi\":\"10.1016/j.crneur.2023.100077\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Charcot-Marie-Tooth disease type 1A (CMT1A) is the most prevalent hereditary demyelinating neuropathy. This autosomal, dominantly inherited disease is caused by a duplication on chromosome 17p which includes the peripheral myelin protein 22 (PMP22) gene. There is clinical evidence that the disability in CMT1A is to a large extend due to axonal damage rather than demyelination. Over-expression of <em>PMP22</em> is recently thought to impede cholesterol trafficking causing a total shutdown of local cholesterol and lipid synthesis in the Schwann cells, thus disturbing their ability to remyelinate. But there is a large variety in disease burden between CMT1A patients with the same genetic defect, indicating the presence of modifying factors that affect disease severity. One of these potential factors is the immune system. Several reports have described patients with co-occurrence of CMT1A with chronic inflammatory demyelinating disease or Guillain-Barré syndrome. We have previously shown in multiple animal models that the innate immune system and specifically the terminal complement system is a driver of inflammatory demyelination. To test the contribution of the terminal complement system to neuroinflammation and disease progression in CMT1A, we inhibited systemic complement C6 in two transgenic mouse models for CMT1A, the C3-<em>PMP22</em> and C3-<em>PMP22</em> c-JunP0Cre models. Both models over-express human <em>PMP22</em>, and one (C3-<em>PMP22</em> c-JunP0Cre) also has a Schwann cell-specific knockout of c-Jun, a crucial regulator of myelination controlling autophagy. We found that systemic inhibition of C6 using antisense oligonucleotides affects the neuroinflammation, Rho GTPase and ERK/MAPK signalling pathways in the CMT1A mouse models. The cholesterol synthesis pathway remained unaffected. Analysis of motor function during treatment with C6 antisense oligonucleotides did not reveal any significant improvement in the CMT1A mouse models. This study shows that the contribution of the terminal complement system to progressive loss of motor function in the CMT1A mouse models tested is limited.</p></div>\",\"PeriodicalId\":72752,\"journal\":{\"name\":\"Current research in neurobiology\",\"volume\":\"4 \",\"pages\":\"Article 100077\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current research in neurobiology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2665945X23000050\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in neurobiology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2665945X23000050","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
The systemic inhibition of the terminal complement system reduces neuroinflammation but does not improve motor function in mouse models of CMT1A with overexpressed PMP22
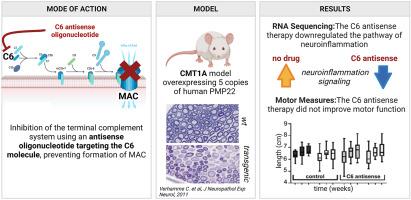
Charcot-Marie-Tooth disease type 1A (CMT1A) is the most prevalent hereditary demyelinating neuropathy. This autosomal, dominantly inherited disease is caused by a duplication on chromosome 17p which includes the peripheral myelin protein 22 (PMP22) gene. There is clinical evidence that the disability in CMT1A is to a large extend due to axonal damage rather than demyelination. Over-expression of PMP22 is recently thought to impede cholesterol trafficking causing a total shutdown of local cholesterol and lipid synthesis in the Schwann cells, thus disturbing their ability to remyelinate. But there is a large variety in disease burden between CMT1A patients with the same genetic defect, indicating the presence of modifying factors that affect disease severity. One of these potential factors is the immune system. Several reports have described patients with co-occurrence of CMT1A with chronic inflammatory demyelinating disease or Guillain-Barré syndrome. We have previously shown in multiple animal models that the innate immune system and specifically the terminal complement system is a driver of inflammatory demyelination. To test the contribution of the terminal complement system to neuroinflammation and disease progression in CMT1A, we inhibited systemic complement C6 in two transgenic mouse models for CMT1A, the C3-PMP22 and C3-PMP22 c-JunP0Cre models. Both models over-express human PMP22, and one (C3-PMP22 c-JunP0Cre) also has a Schwann cell-specific knockout of c-Jun, a crucial regulator of myelination controlling autophagy. We found that systemic inhibition of C6 using antisense oligonucleotides affects the neuroinflammation, Rho GTPase and ERK/MAPK signalling pathways in the CMT1A mouse models. The cholesterol synthesis pathway remained unaffected. Analysis of motor function during treatment with C6 antisense oligonucleotides did not reveal any significant improvement in the CMT1A mouse models. This study shows that the contribution of the terminal complement system to progressive loss of motor function in the CMT1A mouse models tested is limited.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


