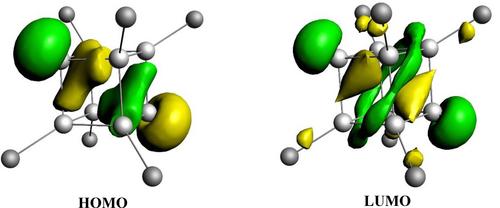
{"title":"八溴化锗Ge8(Sit-butyl2methyl)6中Ge-Ge键的成键分析","authors":"Sudip Pan, Gernot Frenking","doi":"10.1002/ijch.202300062","DOIUrl":null,"url":null,"abstract":"<p>Quantum chemical calculations have been carried out at the BP86/def2-SVP level on Ge<sub>8</sub>(Si<i>t</i>-butyl<sub>2</sub>methyl)<sub>6</sub> (<b>1</b>) and the bonding situation has been analyzed with a variety of methods. The calculated equilibrium geometry of <b>1</b> is in good agreement with the reported x-ray structure analysis. The D3 correction for dispersion interactions as a sum of pairwise attractions leads to an overestimate of the effect of dispersion forces. Calculations at BP86-D3(BJ)/def2-SVP give shorter bonds for Ge(I)−Ge(I) than for Ge(0)−Ge(I), which is in contrast to the experimental values and the BP86/def2-SVP results. The NBO analysis suggests that the best Lewis structure of <b>1</b> has lone-pair orbitals at the Ge(0) atoms with occupation numbers of 1.70 e. A lone-pair character at Ge(0) albeit with less weight is also suggested by the shape of the HOMO, which is an antibonding orbital between the Ge(0) atoms with small contributions from the Ge(I) atoms. The LUMO of <b>1</b> is the corresponding bonding combination of the Ge(0) AOs, which can be explained with the reluctance of the heavier main-group atoms to s/p hybridization of the valence orbitals. The calculated bond order values suggest significant direct Ge(0)−Ge(0) interactions. This is supported by the shape of the HOMO and by the results of EDA-NOCV calculations. The deformation densities and the orbitals associated with the pairwise orbital interaction show that there is a direct charge flow between the Ge(0) atoms of the two fragments, but it is not completely separated from the Ge(0)−Ge(I) and Ge(I)−Ge(I) bond formation. The QTAIM calculations suggest that <b>1</b> has a cubic structure with a cage critical point but not a bond critical point for the Ge(0)−Ge(0) interactions. The dispersion interactions of the large substituents in <b>1</b> have a significant influence on the stability of the compound.</p>","PeriodicalId":14686,"journal":{"name":"Israel Journal of Chemistry","volume":"63 7-8","pages":""},"PeriodicalIF":2.3000,"publicationDate":"2023-07-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ijch.202300062","citationCount":"0","resultStr":"{\"title\":\"Bonding Analysis of the Ge-Ge Bonds in the Octagermacubane Ge8(Sit-butyl2methyl)6\",\"authors\":\"Sudip Pan, Gernot Frenking\",\"doi\":\"10.1002/ijch.202300062\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Quantum chemical calculations have been carried out at the BP86/def2-SVP level on Ge<sub>8</sub>(Si<i>t</i>-butyl<sub>2</sub>methyl)<sub>6</sub> (<b>1</b>) and the bonding situation has been analyzed with a variety of methods. The calculated equilibrium geometry of <b>1</b> is in good agreement with the reported x-ray structure analysis. The D3 correction for dispersion interactions as a sum of pairwise attractions leads to an overestimate of the effect of dispersion forces. Calculations at BP86-D3(BJ)/def2-SVP give shorter bonds for Ge(I)−Ge(I) than for Ge(0)−Ge(I), which is in contrast to the experimental values and the BP86/def2-SVP results. The NBO analysis suggests that the best Lewis structure of <b>1</b> has lone-pair orbitals at the Ge(0) atoms with occupation numbers of 1.70 e. A lone-pair character at Ge(0) albeit with less weight is also suggested by the shape of the HOMO, which is an antibonding orbital between the Ge(0) atoms with small contributions from the Ge(I) atoms. The LUMO of <b>1</b> is the corresponding bonding combination of the Ge(0) AOs, which can be explained with the reluctance of the heavier main-group atoms to s/p hybridization of the valence orbitals. The calculated bond order values suggest significant direct Ge(0)−Ge(0) interactions. This is supported by the shape of the HOMO and by the results of EDA-NOCV calculations. The deformation densities and the orbitals associated with the pairwise orbital interaction show that there is a direct charge flow between the Ge(0) atoms of the two fragments, but it is not completely separated from the Ge(0)−Ge(I) and Ge(I)−Ge(I) bond formation. The QTAIM calculations suggest that <b>1</b> has a cubic structure with a cage critical point but not a bond critical point for the Ge(0)−Ge(0) interactions. The dispersion interactions of the large substituents in <b>1</b> have a significant influence on the stability of the compound.</p>\",\"PeriodicalId\":14686,\"journal\":{\"name\":\"Israel Journal of Chemistry\",\"volume\":\"63 7-8\",\"pages\":\"\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2023-07-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ijch.202300062\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Israel Journal of Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/ijch.202300062\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Israel Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ijch.202300062","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Bonding Analysis of the Ge-Ge Bonds in the Octagermacubane Ge8(Sit-butyl2methyl)6
Quantum chemical calculations have been carried out at the BP86/def2-SVP level on Ge8(Sit-butyl2methyl)6 (1) and the bonding situation has been analyzed with a variety of methods. The calculated equilibrium geometry of 1 is in good agreement with the reported x-ray structure analysis. The D3 correction for dispersion interactions as a sum of pairwise attractions leads to an overestimate of the effect of dispersion forces. Calculations at BP86-D3(BJ)/def2-SVP give shorter bonds for Ge(I)−Ge(I) than for Ge(0)−Ge(I), which is in contrast to the experimental values and the BP86/def2-SVP results. The NBO analysis suggests that the best Lewis structure of 1 has lone-pair orbitals at the Ge(0) atoms with occupation numbers of 1.70 e. A lone-pair character at Ge(0) albeit with less weight is also suggested by the shape of the HOMO, which is an antibonding orbital between the Ge(0) atoms with small contributions from the Ge(I) atoms. The LUMO of 1 is the corresponding bonding combination of the Ge(0) AOs, which can be explained with the reluctance of the heavier main-group atoms to s/p hybridization of the valence orbitals. The calculated bond order values suggest significant direct Ge(0)−Ge(0) interactions. This is supported by the shape of the HOMO and by the results of EDA-NOCV calculations. The deformation densities and the orbitals associated with the pairwise orbital interaction show that there is a direct charge flow between the Ge(0) atoms of the two fragments, but it is not completely separated from the Ge(0)−Ge(I) and Ge(I)−Ge(I) bond formation. The QTAIM calculations suggest that 1 has a cubic structure with a cage critical point but not a bond critical point for the Ge(0)−Ge(0) interactions. The dispersion interactions of the large substituents in 1 have a significant influence on the stability of the compound.
期刊介绍:
The fledgling State of Israel began to publish its scientific activity in 1951 under the general heading of Bulletin of the Research Council of Israel, which quickly split into sections to accommodate various fields in the growing academic community. In 1963, the Bulletin ceased publication and independent journals were born, with Section A becoming the new Israel Journal of Chemistry.
The Israel Journal of Chemistry is the official journal of the Israel Chemical Society. Effective from Volume 50 (2010) it is published by Wiley-VCH.
The Israel Journal of Chemistry is an international and peer-reviewed publication forum for Special Issues on timely research topics in all fields of chemistry: from biochemistry through organic and inorganic chemistry to polymer, physical and theoretical chemistry, including all interdisciplinary topics. Each topical issue is edited by one or several Guest Editors and primarily contains invited Review articles. Communications and Full Papers may be published occasionally, if they fit with the quality standards of the journal. The publication language is English and the journal is published twelve times a year.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


