Jacob Crouch, Tong Mou, Gengnan Li, Daniel Resasco and Bin Wang*,
{"title":"范德华近似方法如何影响Pd表面环己烯加氢的激活势垒","authors":"Jacob Crouch, Tong Mou, Gengnan Li, Daniel Resasco and Bin Wang*, ","doi":"10.1021/acsengineeringau.2c00031","DOIUrl":null,"url":null,"abstract":"<p >Inclusion of van der Waals (vdW) interactions in density functional theory (DFT) calculations improves the accuracy of the calculations of molecular structures, solid structures, and molecular adsorption configuration and energy. However, it remains unclear how vdW approximations affect calculations of activation barriers of surface reactions, which is valuable for evaluating the reaction kinetics. In this work, we choose a prototype reaction─cyclohexene hydrogenation on a Pd surface─as an example to compare different approaches to include vdW interactions in the calculation of activation barriers of surface elementary steps. We find that the adsorption of cyclohexene and desorption of the product, cyclohexane, are very sensitive to the approaches used to incorporate vdW interactions, while the intrinsic barrier of hydrogenation only varies by about 10%. As a result, the apparent activation barrier also varies to a large extent (from −1.90 to 0.28 eV). The rate-determining transition state was found to be the first hydrogenation step, independent of the vdW approximation used. These calculations indicate that the comparison of intrinsic (true) activation barriers between experimentally measured activation barriers and calculated values is more straightforward, while the comparison for the apparent activation energy may be less reliable. Therefore, simultaneous measurement of intrinsic and apparent activation barriers could serve as a potential way to benchmark the most reliable vdW approximation for molecular adsorption and reaction.</p>","PeriodicalId":29804,"journal":{"name":"ACS Engineering Au","volume":"2 6","pages":"547–552"},"PeriodicalIF":5.1000,"publicationDate":"2022-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acsengineeringau.2c00031","citationCount":"0","resultStr":"{\"title\":\"How van der Waals Approximation Methods Affect Activation Barriers of Cyclohexene Hydrogenation over a Pd Surface\",\"authors\":\"Jacob Crouch, Tong Mou, Gengnan Li, Daniel Resasco and Bin Wang*, \",\"doi\":\"10.1021/acsengineeringau.2c00031\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Inclusion of van der Waals (vdW) interactions in density functional theory (DFT) calculations improves the accuracy of the calculations of molecular structures, solid structures, and molecular adsorption configuration and energy. However, it remains unclear how vdW approximations affect calculations of activation barriers of surface reactions, which is valuable for evaluating the reaction kinetics. In this work, we choose a prototype reaction─cyclohexene hydrogenation on a Pd surface─as an example to compare different approaches to include vdW interactions in the calculation of activation barriers of surface elementary steps. We find that the adsorption of cyclohexene and desorption of the product, cyclohexane, are very sensitive to the approaches used to incorporate vdW interactions, while the intrinsic barrier of hydrogenation only varies by about 10%. As a result, the apparent activation barrier also varies to a large extent (from −1.90 to 0.28 eV). The rate-determining transition state was found to be the first hydrogenation step, independent of the vdW approximation used. These calculations indicate that the comparison of intrinsic (true) activation barriers between experimentally measured activation barriers and calculated values is more straightforward, while the comparison for the apparent activation energy may be less reliable. Therefore, simultaneous measurement of intrinsic and apparent activation barriers could serve as a potential way to benchmark the most reliable vdW approximation for molecular adsorption and reaction.</p>\",\"PeriodicalId\":29804,\"journal\":{\"name\":\"ACS Engineering Au\",\"volume\":\"2 6\",\"pages\":\"547–552\"},\"PeriodicalIF\":5.1000,\"publicationDate\":\"2022-09-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acsengineeringau.2c00031\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Engineering Au\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acsengineeringau.2c00031\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ENGINEERING, CHEMICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Engineering Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsengineeringau.2c00031","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENGINEERING, CHEMICAL","Score":null,"Total":0}
How van der Waals Approximation Methods Affect Activation Barriers of Cyclohexene Hydrogenation over a Pd Surface
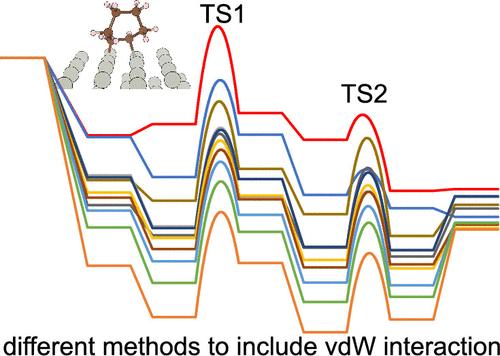
Inclusion of van der Waals (vdW) interactions in density functional theory (DFT) calculations improves the accuracy of the calculations of molecular structures, solid structures, and molecular adsorption configuration and energy. However, it remains unclear how vdW approximations affect calculations of activation barriers of surface reactions, which is valuable for evaluating the reaction kinetics. In this work, we choose a prototype reaction─cyclohexene hydrogenation on a Pd surface─as an example to compare different approaches to include vdW interactions in the calculation of activation barriers of surface elementary steps. We find that the adsorption of cyclohexene and desorption of the product, cyclohexane, are very sensitive to the approaches used to incorporate vdW interactions, while the intrinsic barrier of hydrogenation only varies by about 10%. As a result, the apparent activation barrier also varies to a large extent (from −1.90 to 0.28 eV). The rate-determining transition state was found to be the first hydrogenation step, independent of the vdW approximation used. These calculations indicate that the comparison of intrinsic (true) activation barriers between experimentally measured activation barriers and calculated values is more straightforward, while the comparison for the apparent activation energy may be less reliable. Therefore, simultaneous measurement of intrinsic and apparent activation barriers could serve as a potential way to benchmark the most reliable vdW approximation for molecular adsorption and reaction.
期刊介绍:
)ACS Engineering Au is an open access journal that reports significant advances in chemical engineering applied chemistry and energy covering fundamentals processes and products. The journal's broad scope includes experimental theoretical mathematical computational chemical and physical research from academic and industrial settings. Short letters comprehensive articles reviews and perspectives are welcome on topics that include:Fundamental research in such areas as thermodynamics transport phenomena (flow mixing mass & heat transfer) chemical reaction kinetics and engineering catalysis separations interfacial phenomena and materialsProcess design development and intensification (e.g. process technologies for chemicals and materials synthesis and design methods process intensification multiphase reactors scale-up systems analysis process control data correlation schemes modeling machine learning Artificial Intelligence)Product research and development involving chemical and engineering aspects (e.g. catalysts plastics elastomers fibers adhesives coatings paper membranes lubricants ceramics aerosols fluidic devices intensified process equipment)Energy and fuels (e.g. pre-treatment processing and utilization of renewable energy resources; processing and utilization of fuels; properties and structure or molecular composition of both raw fuels and refined products; fuel cells hydrogen batteries; photochemical fuel and energy production; decarbonization; electrification; microwave; cavitation)Measurement techniques computational models and data on thermo-physical thermodynamic and transport properties of materials and phase equilibrium behaviorNew methods models and tools (e.g. real-time data analytics multi-scale models physics informed machine learning models machine learning enhanced physics-based models soft sensors high-performance computing)

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


