Zhihengyu Chen, Michelle L. Beauvais, Karena W. Chapman
{"title":"PDFgui中离散纳米材料的对分布函数分析","authors":"Zhihengyu Chen, Michelle L. Beauvais, Karena W. Chapman","doi":"10.1107/S1600576723000237","DOIUrl":null,"url":null,"abstract":"<p>Pair distribution functions (PDFs) are a leading tool for atomic structure analysis of nanomaterials. However, the most widely used programs for refining atomic structure against PDF data are based on extended crystallographic models, which cannot be applied to discrete, whole nanoparticles. This work describes a straightforward approach to simulate and refine atomistic models of discrete clusters and nanoparticles employing widely used PDF modelling programs such as <i>PDFgui</i> [Farrow <i>et al.</i> (2007). <i>J. Phys. Condens. Matter</i>, <b>19</b>, 335219] that utilize extended crystallographic models. In this approach, the whole particle to be modelled is contained within an expanded, and otherwise empty, unit cell that is sufficiently large to avoid correlations between atoms in neighbouring unit cells over the <i>r</i> range analysed. The PDF of the particle is simulated as a composite using two conventional `phases': one that calculates the atom–atom correlations and one that approximates the local number density. This approach is first validated for large nanoparticles that are well modelled by a conventional shape factor model, and then applied to simulate the PDF of discrete particles and low-dimensional materials (graphene and MXene) and to model the experimental PDF data for single-layer FeS nanosheets. A comparison of this approach with the <i>DiffPy-CMI</i> program [Juhás <i>et al.</i> (2015). <i>Acta Cryst.</i> A<b>71</b>, 562–568], which calculates the PDF of discrete species, shows that the composite modelling approach is equally or more accurate. Example input files for implementing this approach within <i>PDFgui</i> and <i>TOPAS</i> [Coelho (2018). <i>J. Appl. Cryst.</i><b>51</b>, 210–218], and recommendations for selecting model parameters for reliable application of this refinement strategy, are provided.</p>","PeriodicalId":48737,"journal":{"name":"Journal of Applied Crystallography","volume":"56 2","pages":"328-337"},"PeriodicalIF":5.2000,"publicationDate":"2023-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Pair distribution function analysis of discrete nanomaterials in PDFgui\",\"authors\":\"Zhihengyu Chen, Michelle L. Beauvais, Karena W. Chapman\",\"doi\":\"10.1107/S1600576723000237\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Pair distribution functions (PDFs) are a leading tool for atomic structure analysis of nanomaterials. However, the most widely used programs for refining atomic structure against PDF data are based on extended crystallographic models, which cannot be applied to discrete, whole nanoparticles. This work describes a straightforward approach to simulate and refine atomistic models of discrete clusters and nanoparticles employing widely used PDF modelling programs such as <i>PDFgui</i> [Farrow <i>et al.</i> (2007). <i>J. Phys. Condens. Matter</i>, <b>19</b>, 335219] that utilize extended crystallographic models. In this approach, the whole particle to be modelled is contained within an expanded, and otherwise empty, unit cell that is sufficiently large to avoid correlations between atoms in neighbouring unit cells over the <i>r</i> range analysed. The PDF of the particle is simulated as a composite using two conventional `phases': one that calculates the atom–atom correlations and one that approximates the local number density. This approach is first validated for large nanoparticles that are well modelled by a conventional shape factor model, and then applied to simulate the PDF of discrete particles and low-dimensional materials (graphene and MXene) and to model the experimental PDF data for single-layer FeS nanosheets. A comparison of this approach with the <i>DiffPy-CMI</i> program [Juhás <i>et al.</i> (2015). <i>Acta Cryst.</i> A<b>71</b>, 562–568], which calculates the PDF of discrete species, shows that the composite modelling approach is equally or more accurate. Example input files for implementing this approach within <i>PDFgui</i> and <i>TOPAS</i> [Coelho (2018). <i>J. Appl. Cryst.</i><b>51</b>, 210–218], and recommendations for selecting model parameters for reliable application of this refinement strategy, are provided.</p>\",\"PeriodicalId\":48737,\"journal\":{\"name\":\"Journal of Applied Crystallography\",\"volume\":\"56 2\",\"pages\":\"328-337\"},\"PeriodicalIF\":5.2000,\"publicationDate\":\"2023-02-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Applied Crystallography\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1107/S1600576723000237\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Applied Crystallography","FirstCategoryId":"88","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1107/S1600576723000237","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 1
摘要
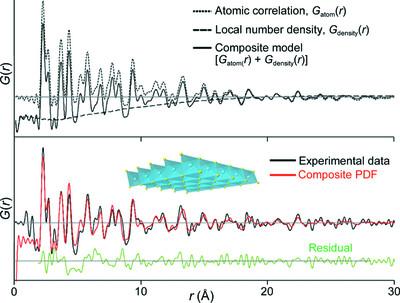
对分布函数(pdf)是纳米材料原子结构分析的主要工具。然而,根据PDF数据提炼原子结构的最广泛使用的程序是基于扩展的晶体学模型,这不能应用于离散的、完整的纳米颗粒。这项工作描述了一种直接的方法来模拟和完善离散簇和纳米颗粒的原子模型,使用广泛使用的PDF建模程序,如PDFgui [Farrow et al.(2007)]。期刊。提供者。物质学报,19(3):357 - 357]。在这种方法中,要建模的整个粒子被包含在一个扩展的,否则是空的,足够大的单元格中,以避免在分析的r范围内相邻单元格中的原子之间的相关性。粒子的PDF是用两个传统的“相”来模拟的:一个计算原子间的相关性,另一个近似局域数密度。该方法首先在传统形状因子模型可以很好地模拟的大型纳米颗粒上得到验证,然后应用于模拟离散颗粒和低维材料(石墨烯和MXene)的PDF,并对单层FeS纳米片的实验PDF数据进行建模。该方法与DiffPy-CMI程序的比较[Juhás et al.(2015)]。Acta结晶。[A71, 562-568],它计算了离散物种的PDF,表明复合建模方法同样或更准确。在PDFgui和TOPAS中实现此方法的示例输入文件[Coelho(2018)]。j:。Cryst. 51, 210-218],并提供了选择模型参数以可靠地应用该优化策略的建议。
Pair distribution function analysis of discrete nanomaterials in PDFgui
Pair distribution functions (PDFs) are a leading tool for atomic structure analysis of nanomaterials. However, the most widely used programs for refining atomic structure against PDF data are based on extended crystallographic models, which cannot be applied to discrete, whole nanoparticles. This work describes a straightforward approach to simulate and refine atomistic models of discrete clusters and nanoparticles employing widely used PDF modelling programs such as PDFgui [Farrow et al. (2007). J. Phys. Condens. Matter, 19, 335219] that utilize extended crystallographic models. In this approach, the whole particle to be modelled is contained within an expanded, and otherwise empty, unit cell that is sufficiently large to avoid correlations between atoms in neighbouring unit cells over the r range analysed. The PDF of the particle is simulated as a composite using two conventional `phases': one that calculates the atom–atom correlations and one that approximates the local number density. This approach is first validated for large nanoparticles that are well modelled by a conventional shape factor model, and then applied to simulate the PDF of discrete particles and low-dimensional materials (graphene and MXene) and to model the experimental PDF data for single-layer FeS nanosheets. A comparison of this approach with the DiffPy-CMI program [Juhás et al. (2015). Acta Cryst. A71, 562–568], which calculates the PDF of discrete species, shows that the composite modelling approach is equally or more accurate. Example input files for implementing this approach within PDFgui and TOPAS [Coelho (2018). J. Appl. Cryst.51, 210–218], and recommendations for selecting model parameters for reliable application of this refinement strategy, are provided.
期刊介绍:
Many research topics in condensed matter research, materials science and the life sciences make use of crystallographic methods to study crystalline and non-crystalline matter with neutrons, X-rays and electrons. Articles published in the Journal of Applied Crystallography focus on these methods and their use in identifying structural and diffusion-controlled phase transformations, structure-property relationships, structural changes of defects, interfaces and surfaces, etc. Developments of instrumentation and crystallographic apparatus, theory and interpretation, numerical analysis and other related subjects are also covered. The journal is the primary place where crystallographic computer program information is published.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


