{"title":"自由能景观的调节作为抗菌肽设计的一种策略","authors":"Sergio A. Hassan, Peter J. Steinbach","doi":"10.1007/s10867-022-09605-z","DOIUrl":null,"url":null,"abstract":"<div><p>Computational design of antimicrobial peptides (AMPs) is a promising area of research for developing novel agents against drug-resistant bacteria. AMPs are present naturally in many organisms, from bacteria to humans, a time-tested mechanism that makes them attractive as effective antibiotics. Depending on the environment, AMPs can exhibit α-helical or β-sheet conformations, a mix of both, or lack secondary structure; they can be linear or cyclic. Prediction of their structures is challenging but critical for rational design. Promising AMP leads can be developed using essentially two approaches: traditional modeling of the physicochemical mechanisms that determine peptide behavior in aqueous and membrane environments and knowledge-based, e.g., machine learning (ML) techniques, that exploit ever-growing AMP databases. Here, we explore the conformational landscapes of two recently ML-designed AMPs, characterize the dependence of these landscapes on the medium conditions, and identify features in peptide and membrane landscapes that mediate protein-membrane association. For both peptides, we observe greater conformational diversity in an aqueous solvent than in a less polar solvent, and one peptide is seen to alter its conformation more dramatically than the other upon the change of solvent. Our results support the view that structural rearrangement in response to environmental changes is central to the mechanism of membrane-structure disruption by linear peptides. We expect that the design of AMPs by ML will benefit from the incorporation of peptide conformational substates as quantified here with molecular simulations.</p></div>","PeriodicalId":612,"journal":{"name":"Journal of Biological Physics","volume":"48 2","pages":"151 - 166"},"PeriodicalIF":2.2000,"publicationDate":"2022-04-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s10867-022-09605-z.pdf","citationCount":"1","resultStr":"{\"title\":\"Modulation of free energy landscapes as a strategy for the design of antimicrobial peptides\",\"authors\":\"Sergio A. Hassan, Peter J. Steinbach\",\"doi\":\"10.1007/s10867-022-09605-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Computational design of antimicrobial peptides (AMPs) is a promising area of research for developing novel agents against drug-resistant bacteria. AMPs are present naturally in many organisms, from bacteria to humans, a time-tested mechanism that makes them attractive as effective antibiotics. Depending on the environment, AMPs can exhibit α-helical or β-sheet conformations, a mix of both, or lack secondary structure; they can be linear or cyclic. Prediction of their structures is challenging but critical for rational design. Promising AMP leads can be developed using essentially two approaches: traditional modeling of the physicochemical mechanisms that determine peptide behavior in aqueous and membrane environments and knowledge-based, e.g., machine learning (ML) techniques, that exploit ever-growing AMP databases. Here, we explore the conformational landscapes of two recently ML-designed AMPs, characterize the dependence of these landscapes on the medium conditions, and identify features in peptide and membrane landscapes that mediate protein-membrane association. For both peptides, we observe greater conformational diversity in an aqueous solvent than in a less polar solvent, and one peptide is seen to alter its conformation more dramatically than the other upon the change of solvent. Our results support the view that structural rearrangement in response to environmental changes is central to the mechanism of membrane-structure disruption by linear peptides. We expect that the design of AMPs by ML will benefit from the incorporation of peptide conformational substates as quantified here with molecular simulations.</p></div>\",\"PeriodicalId\":612,\"journal\":{\"name\":\"Journal of Biological Physics\",\"volume\":\"48 2\",\"pages\":\"151 - 166\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2022-04-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s10867-022-09605-z.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Biological Physics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10867-022-09605-z\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOPHYSICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Biological Physics","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10867-022-09605-z","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOPHYSICS","Score":null,"Total":0}
Modulation of free energy landscapes as a strategy for the design of antimicrobial peptides
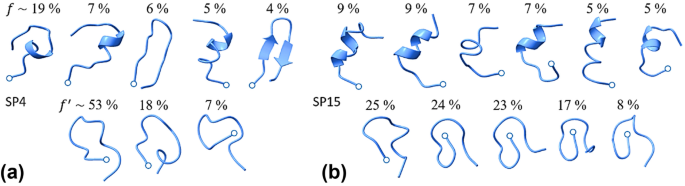
Computational design of antimicrobial peptides (AMPs) is a promising area of research for developing novel agents against drug-resistant bacteria. AMPs are present naturally in many organisms, from bacteria to humans, a time-tested mechanism that makes them attractive as effective antibiotics. Depending on the environment, AMPs can exhibit α-helical or β-sheet conformations, a mix of both, or lack secondary structure; they can be linear or cyclic. Prediction of their structures is challenging but critical for rational design. Promising AMP leads can be developed using essentially two approaches: traditional modeling of the physicochemical mechanisms that determine peptide behavior in aqueous and membrane environments and knowledge-based, e.g., machine learning (ML) techniques, that exploit ever-growing AMP databases. Here, we explore the conformational landscapes of two recently ML-designed AMPs, characterize the dependence of these landscapes on the medium conditions, and identify features in peptide and membrane landscapes that mediate protein-membrane association. For both peptides, we observe greater conformational diversity in an aqueous solvent than in a less polar solvent, and one peptide is seen to alter its conformation more dramatically than the other upon the change of solvent. Our results support the view that structural rearrangement in response to environmental changes is central to the mechanism of membrane-structure disruption by linear peptides. We expect that the design of AMPs by ML will benefit from the incorporation of peptide conformational substates as quantified here with molecular simulations.
期刊介绍:
Many physicists are turning their attention to domains that were not traditionally part of physics and are applying the sophisticated tools of theoretical, computational and experimental physics to investigate biological processes, systems and materials.
The Journal of Biological Physics provides a medium where this growing community of scientists can publish its results and discuss its aims and methods. It welcomes papers which use the tools of physics in an innovative way to study biological problems, as well as research aimed at providing a better understanding of the physical principles underlying biological processes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


