烯的分子内氢酰胺化,通过转位NiH催化实现β-内酰胺的不对称合成
IF 42.8
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
构建对映体β-内酰胺的合成方法非常有价值,因为它们在生物活性化合物中无处不在,尤其是在青霉素和碳青霉烯类等抗生素中。β,γ-不饱和酰胺的分子内氢化反应为进入这一诱人的化学领域提供了便捷的途径,但由于难以形成受约束的四元环而产生的区域选择性问题,这种方法仍然受到限制。在这里,我们介绍了一种镍氢催化的策略,通过使用容易获得的烯基二恶唑酮衍生物来解决这一难题。该反应超越了传统的 NiH 操作模式,通过 N-活化引发的转位机制,从而实现了近端 C-N 键的形成,并具有极佳的区域选择性,与取代基的电子特性无关。这一机制平台对于烯烃的对映体选择性分子内氢化也非常有效,可以方便地获得对映体丰富的 β-内酰胺。β-内酰胺的不对称合成策略备受关注。现在,我们介绍了一种由 NiH 催化的烯烃分子内对映体选择性氢酰胺化反应,从而得到 β-内酰胺支架,其中 C-N 键是在较近的位置以非同寻常的区域选择性形成的。本文章由计算机程序翻译,如有差异,请以英文原文为准。
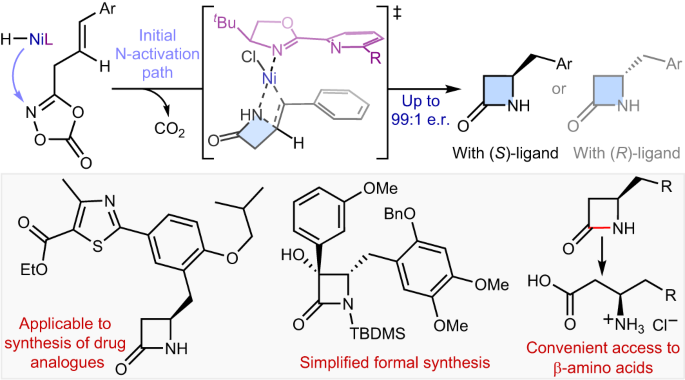
Intramolecular hydroamidation of alkenes enabling asymmetric synthesis of β-lactams via transposed NiH catalysis
Synthetic methods for constructing enantioenriched β-lactams are highly valuable given their ubiquity in bioactive compounds, most notably in antibiotics such as penicillins and carbapenems. Intramolecular hydroamidation of β,γ-unsaturated amides would provide a convenient means to reach this alluring chemical space, yet it remains limited due to the regioselectivity issue arising from the difficulty associated with the formation of strained four-membered rings. Here we describe a NiH-catalysed strategy that addresses this challenge through the use of readily accessible alkenyl dioxazolone derivatives. The reaction transcends the conventional NiH operation mode via a transposed mechanism initiated by N-activation, thus allowing for proximal C–N bond formation with excellent regioselectivity, regardless of the electronic properties of substituents. This mechanistic platform is also highly effective for the enantioselective intramolecular hydroamidation of alkenes to enable a convenient access to enantioenriched β-lactams. Strategies for the asymmetric synthesis of β-lactams are highly sought after. Now, a NiH-catalysed enantioselective intramolecular hydroamidation of alkenes affording β-lactam scaffolds is described, whereby the C–N bond is formed with unusual regioselectivity at the more proximal position.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Catalysis
Chemical Engineering-Bioengineering
CiteScore
52.10
自引率
1.10%
发文量
140
期刊介绍:
Nature Catalysis serves as a platform for researchers across chemistry and related fields, focusing on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, encompassing both fundamental and applied studies. With a particular emphasis on advancing sustainable industries and processes, the journal provides comprehensive coverage of catalysis research, appealing to scientists, engineers, and researchers in academia and industry.
Maintaining the high standards of the Nature brand, Nature Catalysis boasts a dedicated team of professional editors, rigorous peer-review processes, and swift publication times, ensuring editorial independence and quality. The journal publishes work spanning heterogeneous catalysis, homogeneous catalysis, and biocatalysis, covering areas such as catalytic synthesis, mechanisms, characterization, computational studies, nanoparticle catalysis, electrocatalysis, photocatalysis, environmental catalysis, asymmetric catalysis, and various forms of organocatalysis.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


