{"title":"uAUG在引起遗传性出血性毛细血管扩张的ENG 5’UTR中产生变体。","authors":"Omar Soukarieh, Emmanuelle Tillet, Carole Proust, Charlène Dupont, Béatrice Jaspard-Vinassa, Florent Soubrier, Aurélie Goyenvalle, Mélanie Eyries, David-Alexandre Trégouët","doi":"10.1038/s41525-023-00378-5","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary Hemorrhagic Telangiectasia (HHT) is a rare, autosomal dominant, vascular disorder. About 80% of cases are caused by pathogenic variants in ACVRL1 (also known as ALK1) and ENG, with the remaining cases being unexplained. We identified two variants, c.-79C>T and c.-68G>A, in the 5'UTR of ENG in two unrelated patients. They create upstream AUGs at the origin of upstream overlapping open reading frames (uoORFs) ending at the same stop codon. To assess the pathogenicity of these variants, we performed functional assays based on the expression of wild-type and mutant constructs in human cells and evaluated their effect on ALK1 activity in a BMP-response element assay. This assay is mandatory for molecular diagnosis and has been so far only applied to coding ENG variants. These variants were associated with a decrease of protein levels in HeLa and HUVEC cells and a decreased ability to activate ALK1. We applied the same experiments on three additional uoORF-creating variants (c.-142A>T, c.-127C>T and c.-10C>T) located in the 5'UTR of ENG and previously reported in HHT patients. We found that all the analyzed variants alter protein levels and function. Additional experiments relying on an artificial deletion in our mutated constructs show that identified uAUGs could initiate the translation indicating that the associated effect is translation-dependent. Overall, we have identified two 5'UTR ENG variations in HHT patients and shed new light on the role of upstream ORFs on ENG regulation. Our findings contribute to the amelioration of molecular diagnosis in HHT.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"8 1","pages":"32"},"PeriodicalIF":4.7000,"publicationDate":"2023-10-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10582052/pdf/","citationCount":"0","resultStr":"{\"title\":\"uAUG creating variants in the 5'UTR of ENG causing Hereditary Hemorrhagic Telangiectasia.\",\"authors\":\"Omar Soukarieh, Emmanuelle Tillet, Carole Proust, Charlène Dupont, Béatrice Jaspard-Vinassa, Florent Soubrier, Aurélie Goyenvalle, Mélanie Eyries, David-Alexandre Trégouët\",\"doi\":\"10.1038/s41525-023-00378-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hereditary Hemorrhagic Telangiectasia (HHT) is a rare, autosomal dominant, vascular disorder. About 80% of cases are caused by pathogenic variants in ACVRL1 (also known as ALK1) and ENG, with the remaining cases being unexplained. We identified two variants, c.-79C>T and c.-68G>A, in the 5'UTR of ENG in two unrelated patients. They create upstream AUGs at the origin of upstream overlapping open reading frames (uoORFs) ending at the same stop codon. To assess the pathogenicity of these variants, we performed functional assays based on the expression of wild-type and mutant constructs in human cells and evaluated their effect on ALK1 activity in a BMP-response element assay. This assay is mandatory for molecular diagnosis and has been so far only applied to coding ENG variants. These variants were associated with a decrease of protein levels in HeLa and HUVEC cells and a decreased ability to activate ALK1. We applied the same experiments on three additional uoORF-creating variants (c.-142A>T, c.-127C>T and c.-10C>T) located in the 5'UTR of ENG and previously reported in HHT patients. We found that all the analyzed variants alter protein levels and function. Additional experiments relying on an artificial deletion in our mutated constructs show that identified uAUGs could initiate the translation indicating that the associated effect is translation-dependent. Overall, we have identified two 5'UTR ENG variations in HHT patients and shed new light on the role of upstream ORFs on ENG regulation. Our findings contribute to the amelioration of molecular diagnosis in HHT.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"8 1\",\"pages\":\"32\"},\"PeriodicalIF\":4.7000,\"publicationDate\":\"2023-10-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10582052/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-023-00378-5\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00378-5","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
uAUG creating variants in the 5'UTR of ENG causing Hereditary Hemorrhagic Telangiectasia.
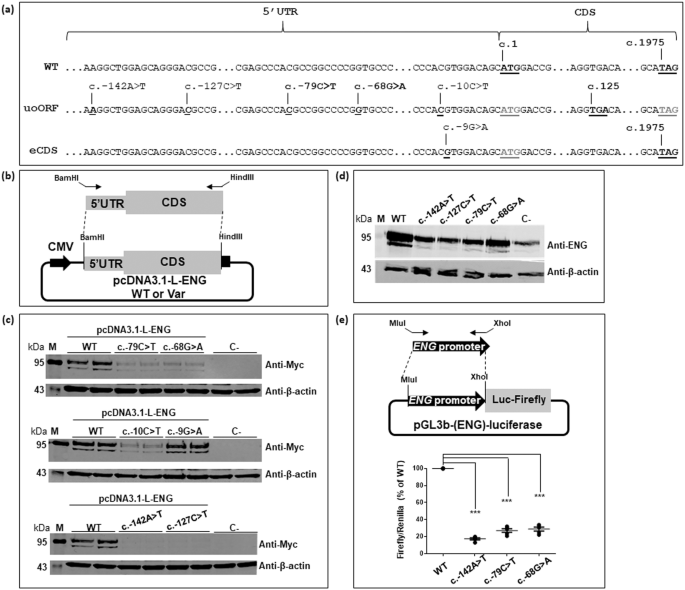
Hereditary Hemorrhagic Telangiectasia (HHT) is a rare, autosomal dominant, vascular disorder. About 80% of cases are caused by pathogenic variants in ACVRL1 (also known as ALK1) and ENG, with the remaining cases being unexplained. We identified two variants, c.-79C>T and c.-68G>A, in the 5'UTR of ENG in two unrelated patients. They create upstream AUGs at the origin of upstream overlapping open reading frames (uoORFs) ending at the same stop codon. To assess the pathogenicity of these variants, we performed functional assays based on the expression of wild-type and mutant constructs in human cells and evaluated their effect on ALK1 activity in a BMP-response element assay. This assay is mandatory for molecular diagnosis and has been so far only applied to coding ENG variants. These variants were associated with a decrease of protein levels in HeLa and HUVEC cells and a decreased ability to activate ALK1. We applied the same experiments on three additional uoORF-creating variants (c.-142A>T, c.-127C>T and c.-10C>T) located in the 5'UTR of ENG and previously reported in HHT patients. We found that all the analyzed variants alter protein levels and function. Additional experiments relying on an artificial deletion in our mutated constructs show that identified uAUGs could initiate the translation indicating that the associated effect is translation-dependent. Overall, we have identified two 5'UTR ENG variations in HHT patients and shed new light on the role of upstream ORFs on ENG regulation. Our findings contribute to the amelioration of molecular diagnosis in HHT.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


