Anna Mucha, Błażej Nowak, Stanisław Dzimira, Bartłomiej Liszka, Magdalena Zatoń-Dobrowolska
{"title":"基于全基因组关联研究的犬乳腺肿瘤SNP标记物鉴定——初步结果。","authors":"Anna Mucha, Błażej Nowak, Stanisław Dzimira, Bartłomiej Liszka, Magdalena Zatoń-Dobrowolska","doi":"10.2478/jvetres-2023-0040","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>The development of genetic research over recent decades has enabled the discovery of new genetic markers, such as single nucleotide polymorphisms (SNPs). This, as well as the full sequencing of the dog genome, has enabled genome-wide association studies (GWAS) to be used in the search for genetic causes of canine mammary tumours (CMTs).</p><p><strong>Material and methods: </strong>Genotypic data containing 175,000 SNPs, which had been obtained using the Illumina CanineHD BeadChip microarray technique, were available for analysis in this study. The data concerned 118 bitches, including 36 animals with CMT, representing various breeds and age groups. Statistical analysis was performed in two steps: quality control of genotyping data and genome-wide association analysis based on dominant, recessive, overdominant, codominant, and log-additive models with the single SNP effects.</p><p><strong>Results: </strong>A total of 40 different SNPs significantly associated with CMT appearance were detected. Moreover, twelve SNPs showed statistical significance in more than one model. Of all the significant SNPs, two, namely <i>BICF2G630136001</i> in the overdominant model and <i>TIGRP2P107898_rs9044787</i> in the log-additive model, reached the 5<sup>-8</sup> significance level. The other SNPs were significant to a 1<sup>-5</sup> level.</p><p><strong>Conclusion: </strong>In the group of SNPs indicated as significant in the GWAS analysis, several transpired to be localised within genes that may play an important role in CMT.</p>","PeriodicalId":17617,"journal":{"name":"Journal of Veterinary Research","volume":"67 3","pages":"427-436"},"PeriodicalIF":1.3000,"publicationDate":"2023-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f5/79/jvetres-67-3-jvetres-2023-0040.PMC10541661.pdf","citationCount":"1","resultStr":"{\"title\":\"Identification of SNP markers for canine mammary gland tumours in females based on a genome-wide association study - preliminary results.\",\"authors\":\"Anna Mucha, Błażej Nowak, Stanisław Dzimira, Bartłomiej Liszka, Magdalena Zatoń-Dobrowolska\",\"doi\":\"10.2478/jvetres-2023-0040\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>The development of genetic research over recent decades has enabled the discovery of new genetic markers, such as single nucleotide polymorphisms (SNPs). This, as well as the full sequencing of the dog genome, has enabled genome-wide association studies (GWAS) to be used in the search for genetic causes of canine mammary tumours (CMTs).</p><p><strong>Material and methods: </strong>Genotypic data containing 175,000 SNPs, which had been obtained using the Illumina CanineHD BeadChip microarray technique, were available for analysis in this study. The data concerned 118 bitches, including 36 animals with CMT, representing various breeds and age groups. Statistical analysis was performed in two steps: quality control of genotyping data and genome-wide association analysis based on dominant, recessive, overdominant, codominant, and log-additive models with the single SNP effects.</p><p><strong>Results: </strong>A total of 40 different SNPs significantly associated with CMT appearance were detected. Moreover, twelve SNPs showed statistical significance in more than one model. Of all the significant SNPs, two, namely <i>BICF2G630136001</i> in the overdominant model and <i>TIGRP2P107898_rs9044787</i> in the log-additive model, reached the 5<sup>-8</sup> significance level. The other SNPs were significant to a 1<sup>-5</sup> level.</p><p><strong>Conclusion: </strong>In the group of SNPs indicated as significant in the GWAS analysis, several transpired to be localised within genes that may play an important role in CMT.</p>\",\"PeriodicalId\":17617,\"journal\":{\"name\":\"Journal of Veterinary Research\",\"volume\":\"67 3\",\"pages\":\"427-436\"},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2023-09-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f5/79/jvetres-67-3-jvetres-2023-0040.PMC10541661.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Veterinary Research\",\"FirstCategoryId\":\"97\",\"ListUrlMain\":\"https://doi.org/10.2478/jvetres-2023-0040\",\"RegionNum\":3,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"VETERINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Veterinary Research","FirstCategoryId":"97","ListUrlMain":"https://doi.org/10.2478/jvetres-2023-0040","RegionNum":3,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"VETERINARY SCIENCES","Score":null,"Total":0}
Identification of SNP markers for canine mammary gland tumours in females based on a genome-wide association study - preliminary results.
Introduction: The development of genetic research over recent decades has enabled the discovery of new genetic markers, such as single nucleotide polymorphisms (SNPs). This, as well as the full sequencing of the dog genome, has enabled genome-wide association studies (GWAS) to be used in the search for genetic causes of canine mammary tumours (CMTs).
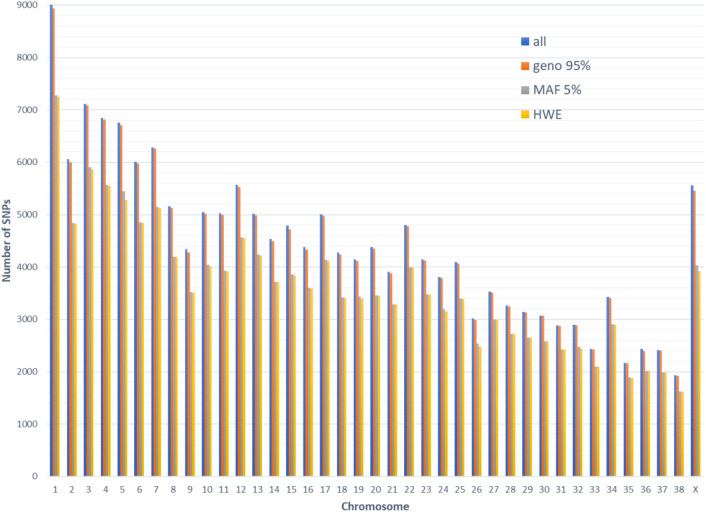
Material and methods: Genotypic data containing 175,000 SNPs, which had been obtained using the Illumina CanineHD BeadChip microarray technique, were available for analysis in this study. The data concerned 118 bitches, including 36 animals with CMT, representing various breeds and age groups. Statistical analysis was performed in two steps: quality control of genotyping data and genome-wide association analysis based on dominant, recessive, overdominant, codominant, and log-additive models with the single SNP effects.
Results: A total of 40 different SNPs significantly associated with CMT appearance were detected. Moreover, twelve SNPs showed statistical significance in more than one model. Of all the significant SNPs, two, namely BICF2G630136001 in the overdominant model and TIGRP2P107898_rs9044787 in the log-additive model, reached the 5-8 significance level. The other SNPs were significant to a 1-5 level.
Conclusion: In the group of SNPs indicated as significant in the GWAS analysis, several transpired to be localised within genes that may play an important role in CMT.
期刊介绍:
Journal of Veterinary Research (formerly Bulletin of the Veterinary Institute in Pulawy) is a quarterly that publishes original papers, review articles and short communications on bacteriology, virology, parasitology, immunology, molecular biology, pathology, toxicology, pharmacology, and biochemistry. The main emphasis is, however, on infectious diseases of animals, food safety and public health, and clinical sciences.
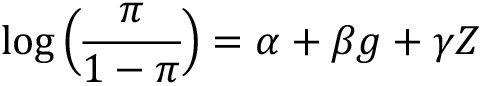

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


