{"title":"单细胞和WGCNA揭示了结直肠癌的预后模型和潜在的致癌基因。","authors":"Ziyang Di, Sicheng Zhou, Gaoran Xu, Lian Ren, Chengxin Li, Zheyu Ding, Kaixin Huang, Leilei Liang, Yihang Yuan","doi":"10.1186/s12575-022-00175-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Colorectal cancer (CRC) is one of the leading causes of cancer-related death worldwide. Single-cell transcriptome sequencing (scRNA-seq) can provide accurate gene expression data for individual cells. In this study, a new prognostic model was constructed by scRNA-seq and bulk transcriptome sequencing (bulk RNA-seq) data of CRC samples to develop a new understanding of CRC.</p><p><strong>Methods: </strong>CRC scRNA-seq data were downloaded from the GSE161277 database, and CRC bulk RNA-seq data were downloaded from the TCGA and GSE17537 databases. The cells were clustered by the FindNeighbors and FindClusters functions in scRNA-seq data. CIBERSORTx was applied to detect the abundance of cell clusters in the bulk RNA-seq expression matrix. WGCNA was performed with the expression profiles to construct the gene coexpression networks of TCGA-CRC. Next, we used a tenfold cross test to construct the model and a nomogram to assess the independence of the model for clinical application. Finally, we examined the expression of the unreported model genes by qPCR and immunohistochemistry. A clone formation assay and orthotopic colorectal tumour model were applied to detect the regulatory roles of unreported model genes.</p><p><strong>Results: </strong>A total of 43,851 cells were included after quality control, and 20 cell clusters were classified by the FindCluster () function. We found that the abundances of C1, C2, C4, C5, C15, C16 and C19 were high and the abundances of C7, C10, C11, C13, C14 and C17 were low in CRC tumour tissues. Meanwhile, the results of survival analysis showed that high abundances of C4, C11 and C13 and low abundances of C5 and C14 were associated with better survival. The WGCNA results showed that the red module was most related to the tumour and the C14 cluster, which contains 615 genes. Lasso Cox regression analysis revealed 8 genes (PBXIP1, MPMZ, SCARA3, INA, ILK, MPP2, L1CAM and FLNA), which were chosen to construct a risk model. In the model, the risk score features had the greatest impact on survival prediction, indicating that the 8-gene risk model can better predict prognosis. qPCR and immunohistochemistry analysis showed that the expression levels of MPZ, SCARA3, MPP2 and PBXIP1 were high in CRC tissues. The functional experiment results indicated that MPZ, SCARA3, MPP2 and PBXIP1 could promote the colony formation ability of CRC cells in vitro and tumorigenicity in vivo.</p><p><strong>Conclusions: </strong>We constructed a risk model to predict the prognosis of CRC patients based on scRNA-seq and bulk RNA-seq data, which could be used for clinical application. We also identified 4 previously unreported model genes (MPZ, SCARA3, MPP2 and PBXIP1) as novel oncogenes in CRC. These results suggest that this model could potentially be used to evaluate the prognostic risk and provide potential therapeutic targets for CRC patients.</p>","PeriodicalId":8960,"journal":{"name":"Biological Procedures Online","volume":" ","pages":"13"},"PeriodicalIF":4.3000,"publicationDate":"2022-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9484253/pdf/","citationCount":"7","resultStr":"{\"title\":\"Single-cell and WGCNA uncover a prognostic model and potential oncogenes in colorectal cancer.\",\"authors\":\"Ziyang Di, Sicheng Zhou, Gaoran Xu, Lian Ren, Chengxin Li, Zheyu Ding, Kaixin Huang, Leilei Liang, Yihang Yuan\",\"doi\":\"10.1186/s12575-022-00175-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Colorectal cancer (CRC) is one of the leading causes of cancer-related death worldwide. Single-cell transcriptome sequencing (scRNA-seq) can provide accurate gene expression data for individual cells. In this study, a new prognostic model was constructed by scRNA-seq and bulk transcriptome sequencing (bulk RNA-seq) data of CRC samples to develop a new understanding of CRC.</p><p><strong>Methods: </strong>CRC scRNA-seq data were downloaded from the GSE161277 database, and CRC bulk RNA-seq data were downloaded from the TCGA and GSE17537 databases. The cells were clustered by the FindNeighbors and FindClusters functions in scRNA-seq data. CIBERSORTx was applied to detect the abundance of cell clusters in the bulk RNA-seq expression matrix. WGCNA was performed with the expression profiles to construct the gene coexpression networks of TCGA-CRC. Next, we used a tenfold cross test to construct the model and a nomogram to assess the independence of the model for clinical application. Finally, we examined the expression of the unreported model genes by qPCR and immunohistochemistry. A clone formation assay and orthotopic colorectal tumour model were applied to detect the regulatory roles of unreported model genes.</p><p><strong>Results: </strong>A total of 43,851 cells were included after quality control, and 20 cell clusters were classified by the FindCluster () function. We found that the abundances of C1, C2, C4, C5, C15, C16 and C19 were high and the abundances of C7, C10, C11, C13, C14 and C17 were low in CRC tumour tissues. Meanwhile, the results of survival analysis showed that high abundances of C4, C11 and C13 and low abundances of C5 and C14 were associated with better survival. The WGCNA results showed that the red module was most related to the tumour and the C14 cluster, which contains 615 genes. Lasso Cox regression analysis revealed 8 genes (PBXIP1, MPMZ, SCARA3, INA, ILK, MPP2, L1CAM and FLNA), which were chosen to construct a risk model. In the model, the risk score features had the greatest impact on survival prediction, indicating that the 8-gene risk model can better predict prognosis. qPCR and immunohistochemistry analysis showed that the expression levels of MPZ, SCARA3, MPP2 and PBXIP1 were high in CRC tissues. The functional experiment results indicated that MPZ, SCARA3, MPP2 and PBXIP1 could promote the colony formation ability of CRC cells in vitro and tumorigenicity in vivo.</p><p><strong>Conclusions: </strong>We constructed a risk model to predict the prognosis of CRC patients based on scRNA-seq and bulk RNA-seq data, which could be used for clinical application. We also identified 4 previously unreported model genes (MPZ, SCARA3, MPP2 and PBXIP1) as novel oncogenes in CRC. These results suggest that this model could potentially be used to evaluate the prognostic risk and provide potential therapeutic targets for CRC patients.</p>\",\"PeriodicalId\":8960,\"journal\":{\"name\":\"Biological Procedures Online\",\"volume\":\" \",\"pages\":\"13\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2022-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9484253/pdf/\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biological Procedures Online\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12575-022-00175-x\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biological Procedures Online","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12575-022-00175-x","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Single-cell and WGCNA uncover a prognostic model and potential oncogenes in colorectal cancer.
Background: Colorectal cancer (CRC) is one of the leading causes of cancer-related death worldwide. Single-cell transcriptome sequencing (scRNA-seq) can provide accurate gene expression data for individual cells. In this study, a new prognostic model was constructed by scRNA-seq and bulk transcriptome sequencing (bulk RNA-seq) data of CRC samples to develop a new understanding of CRC.
Methods: CRC scRNA-seq data were downloaded from the GSE161277 database, and CRC bulk RNA-seq data were downloaded from the TCGA and GSE17537 databases. The cells were clustered by the FindNeighbors and FindClusters functions in scRNA-seq data. CIBERSORTx was applied to detect the abundance of cell clusters in the bulk RNA-seq expression matrix. WGCNA was performed with the expression profiles to construct the gene coexpression networks of TCGA-CRC. Next, we used a tenfold cross test to construct the model and a nomogram to assess the independence of the model for clinical application. Finally, we examined the expression of the unreported model genes by qPCR and immunohistochemistry. A clone formation assay and orthotopic colorectal tumour model were applied to detect the regulatory roles of unreported model genes.
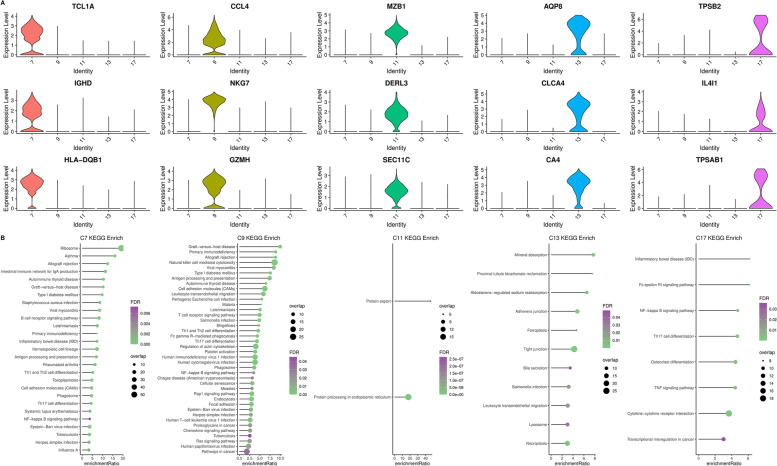
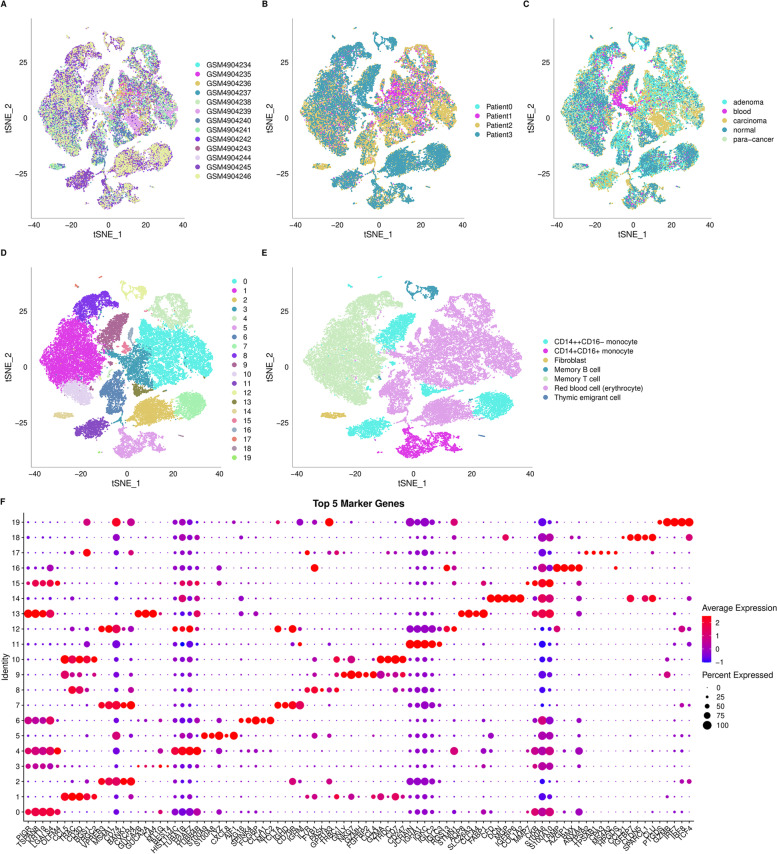
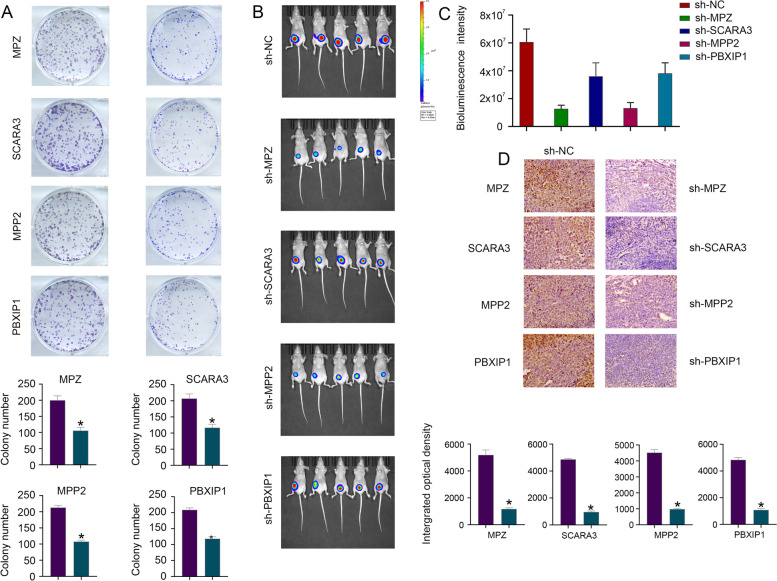
Results: A total of 43,851 cells were included after quality control, and 20 cell clusters were classified by the FindCluster () function. We found that the abundances of C1, C2, C4, C5, C15, C16 and C19 were high and the abundances of C7, C10, C11, C13, C14 and C17 were low in CRC tumour tissues. Meanwhile, the results of survival analysis showed that high abundances of C4, C11 and C13 and low abundances of C5 and C14 were associated with better survival. The WGCNA results showed that the red module was most related to the tumour and the C14 cluster, which contains 615 genes. Lasso Cox regression analysis revealed 8 genes (PBXIP1, MPMZ, SCARA3, INA, ILK, MPP2, L1CAM and FLNA), which were chosen to construct a risk model. In the model, the risk score features had the greatest impact on survival prediction, indicating that the 8-gene risk model can better predict prognosis. qPCR and immunohistochemistry analysis showed that the expression levels of MPZ, SCARA3, MPP2 and PBXIP1 were high in CRC tissues. The functional experiment results indicated that MPZ, SCARA3, MPP2 and PBXIP1 could promote the colony formation ability of CRC cells in vitro and tumorigenicity in vivo.
Conclusions: We constructed a risk model to predict the prognosis of CRC patients based on scRNA-seq and bulk RNA-seq data, which could be used for clinical application. We also identified 4 previously unreported model genes (MPZ, SCARA3, MPP2 and PBXIP1) as novel oncogenes in CRC. These results suggest that this model could potentially be used to evaluate the prognostic risk and provide potential therapeutic targets for CRC patients.
期刊介绍:
iological Procedures Online publishes articles that improve access to techniques and methods in the medical and biological sciences.
We are also interested in short but important research discoveries, such as new animal disease models.
Topics of interest include, but are not limited to:
Reports of new research techniques and applications of existing techniques
Technical analyses of research techniques and published reports
Validity analyses of research methods and approaches to judging the validity of research reports
Application of common research methods
Reviews of existing techniques
Novel/important product information
Biological Procedures Online places emphasis on multidisciplinary approaches that integrate methodologies from medicine, biology, chemistry, imaging, engineering, bioinformatics, computer science, and systems analysis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


