Midhuna Immaculate Joseph Maran, Dicky John Davis G.
{"title":"在预处理环境宏基因组数据之前合并成对端读取的好处","authors":"Midhuna Immaculate Joseph Maran, Dicky John Davis G.","doi":"10.1016/j.margen.2021.100914","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>High throughput sequencing<span><span> of environmental DNA<span> has applications in biodiversity monitoring, taxa abundance estimation, understanding the dynamics of </span></span>community ecology, and marine species studies and conservation. Environmental DNA, especially, marine eDNA, has a fast degradation rate. Aside from the good quality reads, the data could have a significant number of reads that fall slightly below the default PHRED quality threshold of 30 on sequencing. For quality control, trimming methods are employed, which generally precede the merging of the read pairs. However, in the case of eDNA, a significant percentage of reads within the acceptable quality score range are also dropped.</span></p></div><div><h3>Methods</h3><p>To infer the ideal merge tool that is sensitive to eDNA, two Hiseq paired-end eDNA datasets were utilized to study the merging by the tools – FLASH (Fast Length Adjustment of SHort reads), PANDAseq, COPE, BBMerge, and VSEARCH without preprocessing. We assessed these tools on the following parameters: Time taken to process, the quality, and the number of merged reads.</p><p>Trimmomatic, a widely-used preprocessing tool, was also assessed by preprocessing the datasets at different parameters for the two approaches of preprocessing: Sliding Window and Maximum Information. The preprocessed read pairs were then merged using the ideal merge tool identified earlier.</p></div><div><h3>Results</h3><p>FLASH is the most efficient merge tool balancing data conservation, quality of reads, and processing time. We compared Trimmomatic's two quality trimming options with increasing strictness with FLASH's direct merge. The raw reads processed with Trimmomatic then merged, yielded a significant drop in reads compared to the direct merge. An average of 29% of reads was dropped when directly merged with FLASH. Maximum Information option resulted in 30.7% to 68.05% read loss with lowest and highest stringency parameters, respectively. The Sliding Window approach conserves approximately 10% more reads at a PHRED score of 25 set as the threshold for a window of size 4. The lowered PHRED cut off conserves about 50% of the reads that could potentially be informative. We noted no significant reduction of data while optimizing the number of reads read in a window with the ideal quality (Q) score.</p></div><div><h3>Conclusions</h3><p><span>Losing reads can negatively impact the downstream processing of the environmental data, especially for </span>sequence alignment studies. The quality trim-first-merge-later approach can significantly decrease the number of reads conserved. However, direct merging of pair-end reads using FLASH conserved more than 60% of the reads. Therefore, direct merging of the paired-end reads can prevent potential removal of informative reads that do not comply by the trimming tool's strict checks. FLASH to be an efficient tool in conserving reads while carrying out quality trimming in moderation. Overall, our results show that merging paired-end reads of eDNA data before trimming can conserve more reads.</p></div>","PeriodicalId":18321,"journal":{"name":"Marine genomics","volume":"61 ","pages":"Article 100914"},"PeriodicalIF":1.5000,"publicationDate":"2022-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":"{\"title\":\"Benefits of merging paired-end reads before pre-processing environmental metagenomics data\",\"authors\":\"Midhuna Immaculate Joseph Maran, Dicky John Davis G.\",\"doi\":\"10.1016/j.margen.2021.100914\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>High throughput sequencing<span><span> of environmental DNA<span> has applications in biodiversity monitoring, taxa abundance estimation, understanding the dynamics of </span></span>community ecology, and marine species studies and conservation. Environmental DNA, especially, marine eDNA, has a fast degradation rate. Aside from the good quality reads, the data could have a significant number of reads that fall slightly below the default PHRED quality threshold of 30 on sequencing. For quality control, trimming methods are employed, which generally precede the merging of the read pairs. However, in the case of eDNA, a significant percentage of reads within the acceptable quality score range are also dropped.</span></p></div><div><h3>Methods</h3><p>To infer the ideal merge tool that is sensitive to eDNA, two Hiseq paired-end eDNA datasets were utilized to study the merging by the tools – FLASH (Fast Length Adjustment of SHort reads), PANDAseq, COPE, BBMerge, and VSEARCH without preprocessing. We assessed these tools on the following parameters: Time taken to process, the quality, and the number of merged reads.</p><p>Trimmomatic, a widely-used preprocessing tool, was also assessed by preprocessing the datasets at different parameters for the two approaches of preprocessing: Sliding Window and Maximum Information. The preprocessed read pairs were then merged using the ideal merge tool identified earlier.</p></div><div><h3>Results</h3><p>FLASH is the most efficient merge tool balancing data conservation, quality of reads, and processing time. We compared Trimmomatic's two quality trimming options with increasing strictness with FLASH's direct merge. The raw reads processed with Trimmomatic then merged, yielded a significant drop in reads compared to the direct merge. An average of 29% of reads was dropped when directly merged with FLASH. Maximum Information option resulted in 30.7% to 68.05% read loss with lowest and highest stringency parameters, respectively. The Sliding Window approach conserves approximately 10% more reads at a PHRED score of 25 set as the threshold for a window of size 4. The lowered PHRED cut off conserves about 50% of the reads that could potentially be informative. We noted no significant reduction of data while optimizing the number of reads read in a window with the ideal quality (Q) score.</p></div><div><h3>Conclusions</h3><p><span>Losing reads can negatively impact the downstream processing of the environmental data, especially for </span>sequence alignment studies. The quality trim-first-merge-later approach can significantly decrease the number of reads conserved. However, direct merging of pair-end reads using FLASH conserved more than 60% of the reads. Therefore, direct merging of the paired-end reads can prevent potential removal of informative reads that do not comply by the trimming tool's strict checks. FLASH to be an efficient tool in conserving reads while carrying out quality trimming in moderation. Overall, our results show that merging paired-end reads of eDNA data before trimming can conserve more reads.</p></div>\",\"PeriodicalId\":18321,\"journal\":{\"name\":\"Marine genomics\",\"volume\":\"61 \",\"pages\":\"Article 100914\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2022-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Marine genomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1874778721000805\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Marine genomics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1874778721000805","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 3
摘要
环境DNA的高通量测序在生物多样性监测、分类群丰度估算、群落生态动态研究以及海洋物种研究和保护等方面具有重要的应用价值。环境DNA,尤其是海洋DNA,降解速度快。除了高质量的读取之外,数据可能有大量的读取稍微低于默认的PHRED测序质量阈值30。在质量控制方面,采用了修剪方法,通常在合并读对之前进行。然而,在eDNA的情况下,在可接受的质量评分范围内的相当大比例的读数也会下降。方法利用两组Hiseq配对端eDNA数据集,利用FLASH (Fast Length Adjustment of SHort reads)、PANDAseq、COPE、BBMerge和VSEARCH等未进行预处理的工具进行合并研究,以寻找对eDNA敏感的理想合并工具。我们根据以下参数评估这些工具:处理时间、质量和合并读取的数量。采用滑动窗口和最大信息两种预处理方法对不同参数下的数据集进行预处理,评价了常用的预处理工具Trimmomatic。然后使用前面确定的理想合并工具合并预处理的读对。结果flash是平衡数据保存、读取质量和处理时间的最有效的合并工具。我们比较了Trimmomatic的两个质量修剪选项与增加严格与FLASH的直接合并。然后合并Trimmomatic处理的原始读取,与直接合并相比,读取量显著下降。当直接与FLASH合并时,平均有29%的读取被删除。最大信息选项在最低和最高严格参数下的读损失分别为30.7%和68.05%。对于大小为4的窗口,当PHRED分数为25时,滑动窗口方法可以多保存大约10%的读取。降低的PHRED截断值保留了大约50%可能具有信息的读取。我们注意到,在优化具有理想质量(Q)分数的窗口中读取的读取次数时,数据没有显着减少。结论读取丢失会对环境数据的下游处理产生负面影响,特别是对序列比对研究。质量修剪-先合并-后合并的方法可以显著减少保守的读数。然而,使用FLASH直接合并对端读取可以保存60%以上的读取。因此,直接合并成对的末端读段可以防止不符合修剪工具严格检查的信息读段的潜在移除。FLASH是一种有效的工具,在保存读数的同时进行适度的质量修剪。总的来说,我们的研究结果表明,在修剪之前合并eDNA数据的成对末端读取可以节省更多的读取。
Benefits of merging paired-end reads before pre-processing environmental metagenomics data
Background
High throughput sequencing of environmental DNA has applications in biodiversity monitoring, taxa abundance estimation, understanding the dynamics of community ecology, and marine species studies and conservation. Environmental DNA, especially, marine eDNA, has a fast degradation rate. Aside from the good quality reads, the data could have a significant number of reads that fall slightly below the default PHRED quality threshold of 30 on sequencing. For quality control, trimming methods are employed, which generally precede the merging of the read pairs. However, in the case of eDNA, a significant percentage of reads within the acceptable quality score range are also dropped.
Methods
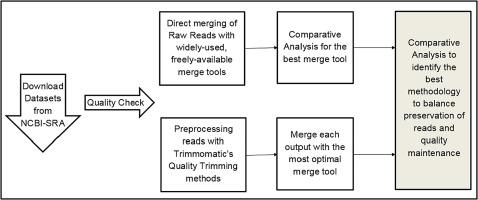
To infer the ideal merge tool that is sensitive to eDNA, two Hiseq paired-end eDNA datasets were utilized to study the merging by the tools – FLASH (Fast Length Adjustment of SHort reads), PANDAseq, COPE, BBMerge, and VSEARCH without preprocessing. We assessed these tools on the following parameters: Time taken to process, the quality, and the number of merged reads.
Trimmomatic, a widely-used preprocessing tool, was also assessed by preprocessing the datasets at different parameters for the two approaches of preprocessing: Sliding Window and Maximum Information. The preprocessed read pairs were then merged using the ideal merge tool identified earlier.
Results
FLASH is the most efficient merge tool balancing data conservation, quality of reads, and processing time. We compared Trimmomatic's two quality trimming options with increasing strictness with FLASH's direct merge. The raw reads processed with Trimmomatic then merged, yielded a significant drop in reads compared to the direct merge. An average of 29% of reads was dropped when directly merged with FLASH. Maximum Information option resulted in 30.7% to 68.05% read loss with lowest and highest stringency parameters, respectively. The Sliding Window approach conserves approximately 10% more reads at a PHRED score of 25 set as the threshold for a window of size 4. The lowered PHRED cut off conserves about 50% of the reads that could potentially be informative. We noted no significant reduction of data while optimizing the number of reads read in a window with the ideal quality (Q) score.
Conclusions
Losing reads can negatively impact the downstream processing of the environmental data, especially for sequence alignment studies. The quality trim-first-merge-later approach can significantly decrease the number of reads conserved. However, direct merging of pair-end reads using FLASH conserved more than 60% of the reads. Therefore, direct merging of the paired-end reads can prevent potential removal of informative reads that do not comply by the trimming tool's strict checks. FLASH to be an efficient tool in conserving reads while carrying out quality trimming in moderation. Overall, our results show that merging paired-end reads of eDNA data before trimming can conserve more reads.
期刊介绍:
The journal publishes papers on all functional and evolutionary aspects of genes, chromatin, chromosomes and (meta)genomes of marine (and freshwater) organisms. It deals with new genome-enabled insights into the broader framework of environmental science. Topics within the scope of this journal include:
• Population genomics and ecology
• Evolutionary and developmental genomics
• Comparative genomics
• Metagenomics
• Environmental genomics
• Systems biology
More specific topics include: geographic and phylogenomic characterization of aquatic organisms, metabolic capacities and pathways of organisms and communities, biogeochemical cycles, genomics and integrative approaches applied to microbial ecology including (meta)transcriptomics and (meta)proteomics, tracking of infectious diseases, environmental stress, global climate change and ecosystem modelling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


