{"title":"预测人类细胞发育过程中与DNA去甲基化相关的转录因子。","authors":"Yurina Miyajima, Shuhei Noguchi, Yuki Tanaka, Jing-Ru Li, Hajime Nishimura, Mami Kishima, Joanne Lim, Erina Furuhata, Takahiro Suzuki, Takeya Kasukawa, Harukazu Suzuki","doi":"10.1007/s10577-022-09685-6","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation of CpG dinucleotides is an important epigenetic modification involved in the regulation of mammalian gene expression, with each type of cell developing a specific methylation profile during its differentiation. Recently, it has been shown that a small subgroup of transcription factors (TFs) might promote DNA demethylation at their binding sites. We developed a bioinformatics pipeline to predict from genome-wide DNA methylation data TFs that promote DNA demethylation at their binding site. We applied the pipeline to International Human Epigenome Consortium methylome data and selected 393 candidate transcription factor binding motifs and associated 383 TFs that are likely associated with DNA demethylation. Validation of a subset of the candidate TFs using an in vitro assay suggested that 28 of 49 TFs from various TF families had DNA-demethylation-promoting activity; TF families, such as bHLH and ETS, contained both TFs with and without the activity. The identified TFs showed large demethylated/methylated CpG ratios and their demethylated CpGs showed significant bias toward hypermethylation in original cells. Furthermore, the identified TFs promoted demethylation of distinct sets of CpGs, with slight overlap of the targeted CpGs among TF family members, which was consistent with the results of a gene ontology (GO) term analysis of the identified TFs. Gene expression analysis of the identified TFs revealed that multiple TFs from various families are specifically expressed in human cells and tissues. Together, our results suggest that a large number of TFs from various TF families are associated with cell-type-specific DNA demethylation during human cellular development.</p>","PeriodicalId":50698,"journal":{"name":"Chromosome Research","volume":"30 1","pages":"109-121"},"PeriodicalIF":2.8000,"publicationDate":"2022-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8942926/pdf/","citationCount":"2","resultStr":"{\"title\":\"Prediction of transcription factors associated with DNA demethylation during human cellular development.\",\"authors\":\"Yurina Miyajima, Shuhei Noguchi, Yuki Tanaka, Jing-Ru Li, Hajime Nishimura, Mami Kishima, Joanne Lim, Erina Furuhata, Takahiro Suzuki, Takeya Kasukawa, Harukazu Suzuki\",\"doi\":\"10.1007/s10577-022-09685-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation of CpG dinucleotides is an important epigenetic modification involved in the regulation of mammalian gene expression, with each type of cell developing a specific methylation profile during its differentiation. Recently, it has been shown that a small subgroup of transcription factors (TFs) might promote DNA demethylation at their binding sites. We developed a bioinformatics pipeline to predict from genome-wide DNA methylation data TFs that promote DNA demethylation at their binding site. We applied the pipeline to International Human Epigenome Consortium methylome data and selected 393 candidate transcription factor binding motifs and associated 383 TFs that are likely associated with DNA demethylation. Validation of a subset of the candidate TFs using an in vitro assay suggested that 28 of 49 TFs from various TF families had DNA-demethylation-promoting activity; TF families, such as bHLH and ETS, contained both TFs with and without the activity. The identified TFs showed large demethylated/methylated CpG ratios and their demethylated CpGs showed significant bias toward hypermethylation in original cells. Furthermore, the identified TFs promoted demethylation of distinct sets of CpGs, with slight overlap of the targeted CpGs among TF family members, which was consistent with the results of a gene ontology (GO) term analysis of the identified TFs. Gene expression analysis of the identified TFs revealed that multiple TFs from various families are specifically expressed in human cells and tissues. Together, our results suggest that a large number of TFs from various TF families are associated with cell-type-specific DNA demethylation during human cellular development.</p>\",\"PeriodicalId\":50698,\"journal\":{\"name\":\"Chromosome Research\",\"volume\":\"30 1\",\"pages\":\"109-121\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2022-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8942926/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Chromosome Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s10577-022-09685-6\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/2/10 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chromosome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s10577-022-09685-6","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/2/10 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Prediction of transcription factors associated with DNA demethylation during human cellular development.
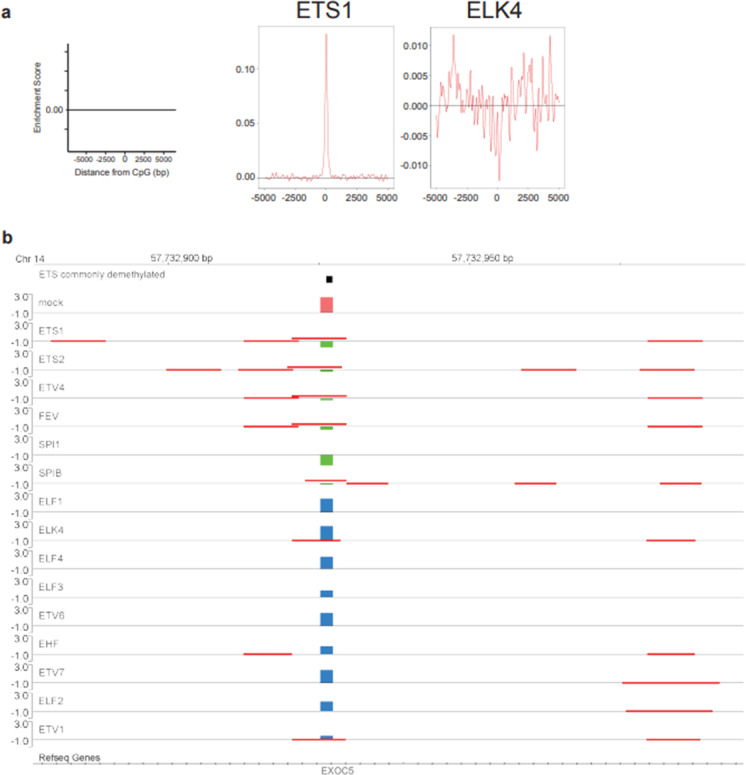
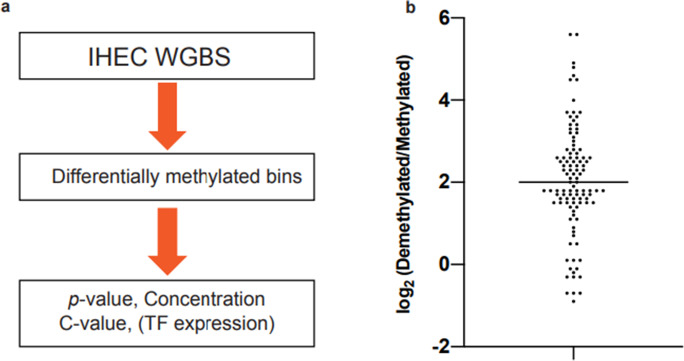
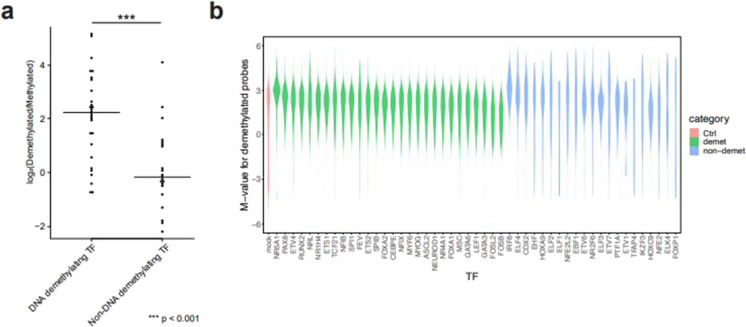
DNA methylation of CpG dinucleotides is an important epigenetic modification involved in the regulation of mammalian gene expression, with each type of cell developing a specific methylation profile during its differentiation. Recently, it has been shown that a small subgroup of transcription factors (TFs) might promote DNA demethylation at their binding sites. We developed a bioinformatics pipeline to predict from genome-wide DNA methylation data TFs that promote DNA demethylation at their binding site. We applied the pipeline to International Human Epigenome Consortium methylome data and selected 393 candidate transcription factor binding motifs and associated 383 TFs that are likely associated with DNA demethylation. Validation of a subset of the candidate TFs using an in vitro assay suggested that 28 of 49 TFs from various TF families had DNA-demethylation-promoting activity; TF families, such as bHLH and ETS, contained both TFs with and without the activity. The identified TFs showed large demethylated/methylated CpG ratios and their demethylated CpGs showed significant bias toward hypermethylation in original cells. Furthermore, the identified TFs promoted demethylation of distinct sets of CpGs, with slight overlap of the targeted CpGs among TF family members, which was consistent with the results of a gene ontology (GO) term analysis of the identified TFs. Gene expression analysis of the identified TFs revealed that multiple TFs from various families are specifically expressed in human cells and tissues. Together, our results suggest that a large number of TFs from various TF families are associated with cell-type-specific DNA demethylation during human cellular development.
期刊介绍:
Chromosome Research publishes manuscripts from work based on all organisms and encourages submissions in the following areas including, but not limited, to:
· Chromosomes and their linkage to diseases;
· Chromosome organization within the nucleus;
· Chromatin biology (transcription, non-coding RNA, etc);
· Chromosome structure, function and mechanics;
· Chromosome and DNA repair;
· Epigenetic chromosomal functions (centromeres, telomeres, replication, imprinting,
dosage compensation, sex determination, chromosome remodeling);
· Architectural/epigenomic organization of the genome;
· Functional annotation of the genome;
· Functional and comparative genomics in plants and animals;
· Karyology studies that help resolve difficult taxonomic problems or that provide
clues to fundamental mechanisms of genome and karyotype evolution in plants and animals;
· Mitosis and Meiosis;
· Cancer cytogenomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


