Junlin Zhang, Yiting Wang, Yingwang Zhao, Fang Liu
{"title":"在一个中国家族中发现的导致常染色体显性多囊肾的 PKD2 非典型剪接突变。","authors":"Junlin Zhang, Yiting Wang, Yingwang Zhao, Fang Liu","doi":"10.11622/smedj.2021162","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Autosomal dominant polycystic kidney disease (ADPKD) is a very common hereditary renal disorder. Mutations in PKD1 and PKD2 , identified as disease-causing genes, account for 85% and 15% of the ADPKD cases, respectively.</p><p><strong>Methods: </strong>In this study, the mutation analysis of polycystic kidney disease (PKD) genes was performed in a Chinese family with suspected ADPKD using targeted clinical exome sequencing (CES). The candidate pathogenic variants were further tested by using Sanger sequencing and validated for co-segregation. In addition, reverse transcription-polymerase chain reaction (RT-PCR) was performed to test for abnormal splicing and assess its potential pathogenicity.</p><p><strong>Results: </strong>A novel atypical splicing mutation that belongs to unclassified variants (UCVs), IVS6+5G>C, was identified in three family members by CES and was shown to co-segregate only with the affected individuals. The RT-PCR revealed the abnormal splicing of exon 6, which thus caused truncating mutation. These findings suggested that the atypical splice site alteration, IVS6+5G>C, in the PKD2 gene was the potential pathogenic mutation leading to ADPKD in this Chinese family.</p><p><strong>Conclusion: </strong>The data available in this study provided strong evidence that IVS6+5G>C is the potential pathogenic mutation for ADPKD. In addition, our findings emphasised the significance of functional analysis of UCVs and genotype-phenotype correlation in ADPKD.</p>","PeriodicalId":21752,"journal":{"name":"Singapore medical journal","volume":" ","pages":"229-234"},"PeriodicalIF":1.9000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11132625/pdf/","citationCount":"0","resultStr":"{\"title\":\"A new atypical splice mutation in PKD2 leading to autosomal dominant polycystic kidney disease in a Chinese family.\",\"authors\":\"Junlin Zhang, Yiting Wang, Yingwang Zhao, Fang Liu\",\"doi\":\"10.11622/smedj.2021162\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Autosomal dominant polycystic kidney disease (ADPKD) is a very common hereditary renal disorder. Mutations in PKD1 and PKD2 , identified as disease-causing genes, account for 85% and 15% of the ADPKD cases, respectively.</p><p><strong>Methods: </strong>In this study, the mutation analysis of polycystic kidney disease (PKD) genes was performed in a Chinese family with suspected ADPKD using targeted clinical exome sequencing (CES). The candidate pathogenic variants were further tested by using Sanger sequencing and validated for co-segregation. In addition, reverse transcription-polymerase chain reaction (RT-PCR) was performed to test for abnormal splicing and assess its potential pathogenicity.</p><p><strong>Results: </strong>A novel atypical splicing mutation that belongs to unclassified variants (UCVs), IVS6+5G>C, was identified in three family members by CES and was shown to co-segregate only with the affected individuals. The RT-PCR revealed the abnormal splicing of exon 6, which thus caused truncating mutation. These findings suggested that the atypical splice site alteration, IVS6+5G>C, in the PKD2 gene was the potential pathogenic mutation leading to ADPKD in this Chinese family.</p><p><strong>Conclusion: </strong>The data available in this study provided strong evidence that IVS6+5G>C is the potential pathogenic mutation for ADPKD. In addition, our findings emphasised the significance of functional analysis of UCVs and genotype-phenotype correlation in ADPKD.</p>\",\"PeriodicalId\":21752,\"journal\":{\"name\":\"Singapore medical journal\",\"volume\":\" \",\"pages\":\"229-234\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2024-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11132625/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Singapore medical journal\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.11622/smedj.2021162\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/11/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Singapore medical journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.11622/smedj.2021162","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/11/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
A new atypical splice mutation in PKD2 leading to autosomal dominant polycystic kidney disease in a Chinese family.
Introduction: Autosomal dominant polycystic kidney disease (ADPKD) is a very common hereditary renal disorder. Mutations in PKD1 and PKD2 , identified as disease-causing genes, account for 85% and 15% of the ADPKD cases, respectively.
Methods: In this study, the mutation analysis of polycystic kidney disease (PKD) genes was performed in a Chinese family with suspected ADPKD using targeted clinical exome sequencing (CES). The candidate pathogenic variants were further tested by using Sanger sequencing and validated for co-segregation. In addition, reverse transcription-polymerase chain reaction (RT-PCR) was performed to test for abnormal splicing and assess its potential pathogenicity.
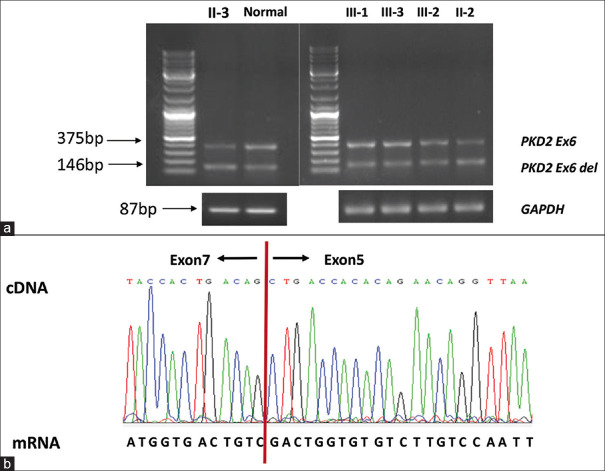
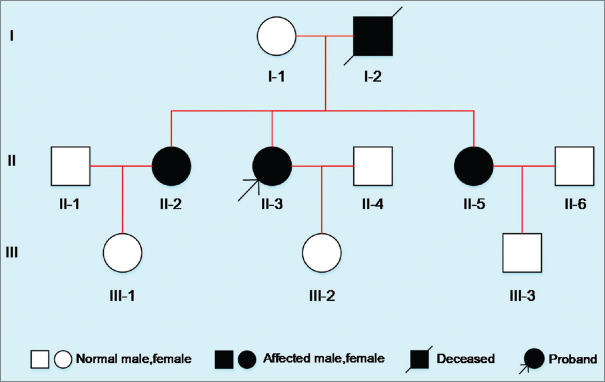
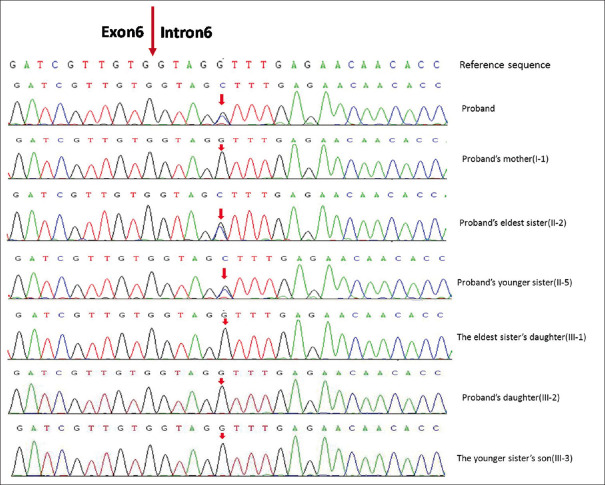
Results: A novel atypical splicing mutation that belongs to unclassified variants (UCVs), IVS6+5G>C, was identified in three family members by CES and was shown to co-segregate only with the affected individuals. The RT-PCR revealed the abnormal splicing of exon 6, which thus caused truncating mutation. These findings suggested that the atypical splice site alteration, IVS6+5G>C, in the PKD2 gene was the potential pathogenic mutation leading to ADPKD in this Chinese family.
Conclusion: The data available in this study provided strong evidence that IVS6+5G>C is the potential pathogenic mutation for ADPKD. In addition, our findings emphasised the significance of functional analysis of UCVs and genotype-phenotype correlation in ADPKD.
期刊介绍:
The Singapore Medical Journal (SMJ) is the monthly publication of Singapore Medical Association (SMA). The Journal aims to advance medical practice and clinical research by publishing high-quality articles that add to the clinical knowledge of physicians in Singapore and worldwide.
SMJ is a general medical journal that focuses on all aspects of human health. The Journal publishes commissioned reviews, commentaries and editorials, original research, a small number of outstanding case reports, continuing medical education articles (ECG Series, Clinics in Diagnostic Imaging, Pictorial Essays, Practice Integration & Life-long Learning [PILL] Series), and short communications in the form of letters to the editor.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


