Liang Tang, Xuezhu Liao, Luke R Tembrock, Song Ge, Zhiqiang Wu
{"title":"濒危热带树Vatica mangachapoi的染色体尺度基因组和转录组学分析。","authors":"Liang Tang, Xuezhu Liao, Luke R Tembrock, Song Ge, Zhiqiang Wu","doi":"10.1093/dnares/dsac005","DOIUrl":null,"url":null,"abstract":"<p><p>Vatica mangachapoi is a tropical tree species native to Southeast Asia. It has long been valued as a timber species because the wood resists decay, but it is now considered vulnerable to extinction due to habitat loss and overexploitation. Here, we present the first chromosome-level genome assembly of V. mangachapoi that we created by combining data from PacBio long read sequencing with Hi-C proximity ligation and Illumina short-read sequencing. The assembled genome was 456.21 Mb, containing 11 chromosome and a BUSCO score of 93.4%. From the newly assembled genome, 46,811 protein-coding genes were predicted. Repetitive DNA accounted for 53% of the genome. Phylogenomic and gene family analyses showed that V. mangachapoi diverged from a common ancestor of Gossypium raimondii 70 million years ago. Transcriptome analyses found 227 genes that were differentially expressed in the leaves of plants grown in normal soil relative to plants grown in dry, coastal, sandy soil. For these genes, we identified three significantly enriched with GO terms: responses to organonitrogen compounds, chitin-triggered immunity, and wound response. This genome provides an important comparative benchmark not only for future conservation work on V. mangachapoi but also for phylogenomics work on Dipterocarpaceae.</p>","PeriodicalId":51014,"journal":{"name":"DNA Research","volume":"29 2","pages":""},"PeriodicalIF":3.9000,"publicationDate":"2022-02-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8882376/pdf/","citationCount":"2","resultStr":"{\"title\":\"A chromosome-scale genome and transcriptomic analysis of the endangered tropical tree Vatica mangachapoi (Dipterocarpaceae).\",\"authors\":\"Liang Tang, Xuezhu Liao, Luke R Tembrock, Song Ge, Zhiqiang Wu\",\"doi\":\"10.1093/dnares/dsac005\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Vatica mangachapoi is a tropical tree species native to Southeast Asia. It has long been valued as a timber species because the wood resists decay, but it is now considered vulnerable to extinction due to habitat loss and overexploitation. Here, we present the first chromosome-level genome assembly of V. mangachapoi that we created by combining data from PacBio long read sequencing with Hi-C proximity ligation and Illumina short-read sequencing. The assembled genome was 456.21 Mb, containing 11 chromosome and a BUSCO score of 93.4%. From the newly assembled genome, 46,811 protein-coding genes were predicted. Repetitive DNA accounted for 53% of the genome. Phylogenomic and gene family analyses showed that V. mangachapoi diverged from a common ancestor of Gossypium raimondii 70 million years ago. Transcriptome analyses found 227 genes that were differentially expressed in the leaves of plants grown in normal soil relative to plants grown in dry, coastal, sandy soil. For these genes, we identified three significantly enriched with GO terms: responses to organonitrogen compounds, chitin-triggered immunity, and wound response. This genome provides an important comparative benchmark not only for future conservation work on V. mangachapoi but also for phylogenomics work on Dipterocarpaceae.</p>\",\"PeriodicalId\":51014,\"journal\":{\"name\":\"DNA Research\",\"volume\":\"29 2\",\"pages\":\"\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2022-02-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8882376/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"DNA Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/dnares/dsac005\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"DNA Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/dnares/dsac005","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
A chromosome-scale genome and transcriptomic analysis of the endangered tropical tree Vatica mangachapoi (Dipterocarpaceae).
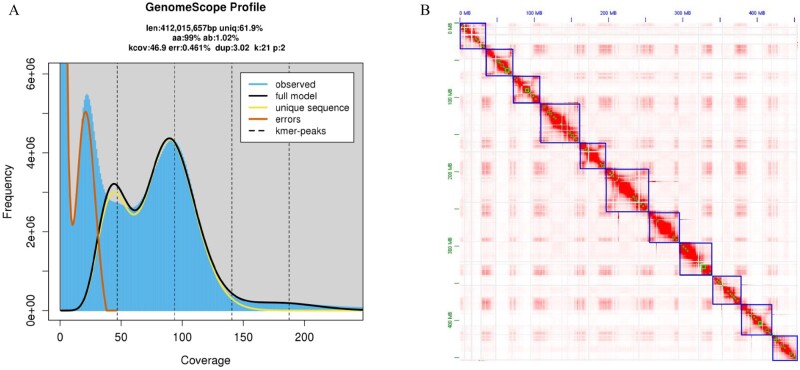
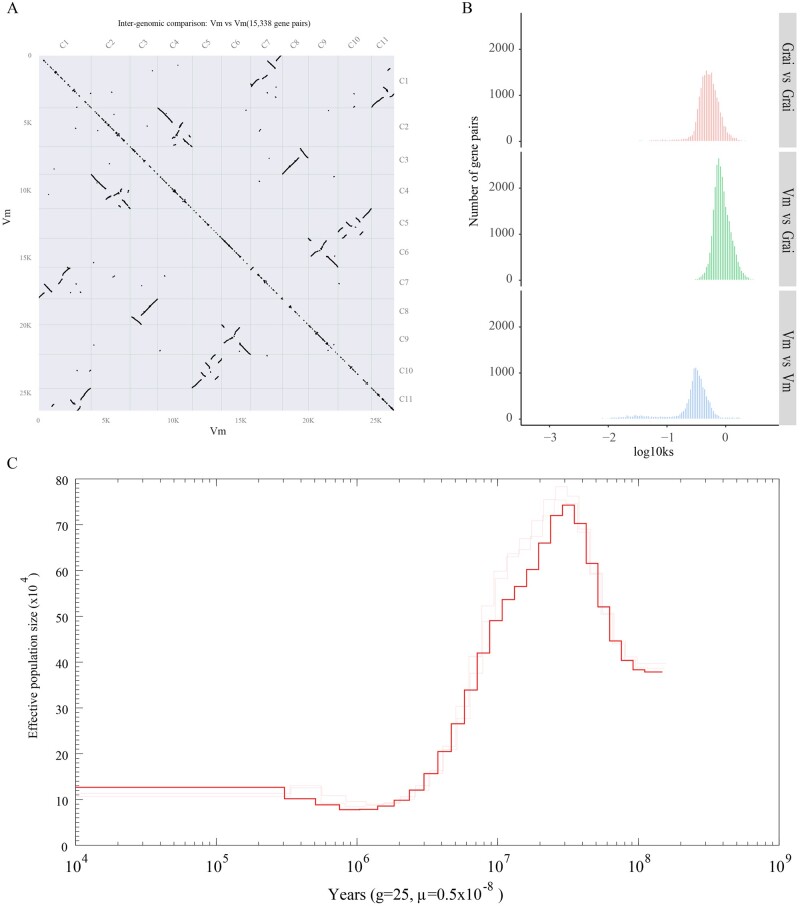
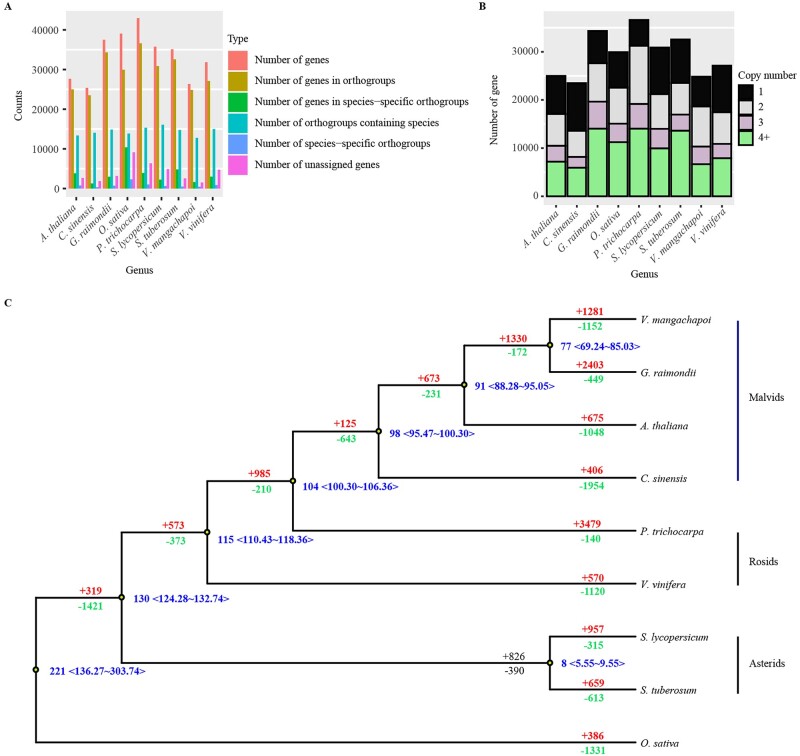
Vatica mangachapoi is a tropical tree species native to Southeast Asia. It has long been valued as a timber species because the wood resists decay, but it is now considered vulnerable to extinction due to habitat loss and overexploitation. Here, we present the first chromosome-level genome assembly of V. mangachapoi that we created by combining data from PacBio long read sequencing with Hi-C proximity ligation and Illumina short-read sequencing. The assembled genome was 456.21 Mb, containing 11 chromosome and a BUSCO score of 93.4%. From the newly assembled genome, 46,811 protein-coding genes were predicted. Repetitive DNA accounted for 53% of the genome. Phylogenomic and gene family analyses showed that V. mangachapoi diverged from a common ancestor of Gossypium raimondii 70 million years ago. Transcriptome analyses found 227 genes that were differentially expressed in the leaves of plants grown in normal soil relative to plants grown in dry, coastal, sandy soil. For these genes, we identified three significantly enriched with GO terms: responses to organonitrogen compounds, chitin-triggered immunity, and wound response. This genome provides an important comparative benchmark not only for future conservation work on V. mangachapoi but also for phylogenomics work on Dipterocarpaceae.
期刊介绍:
DNA Research is an internationally peer-reviewed journal which aims at publishing papers of highest quality in broad aspects of DNA and genome-related research. Emphasis will be made on the following subjects: 1) Sequencing and characterization of genomes/important genomic regions, 2) Comprehensive analysis of the functions of genes, gene families and genomes, 3) Techniques and equipments useful for structural and functional analysis of genes, gene families and genomes, 4) Computer algorithms and/or their applications relevant to structural and functional analysis of genes and genomes. The journal also welcomes novel findings in other scientific disciplines related to genomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


