Moses E Ekpenyong, Anthony A Adegoke, Mercy E Edoho, Udoinyang G Inyang, Ifiok J Udo, Itemobong S Ekaidem, Francis Osang, Nseobong P Uto, Joseph I Geoffery
{"title":"协同挖掘全基因组序列用于智能发现HIV-1亚毒株。","authors":"Moses E Ekpenyong, Anthony A Adegoke, Mercy E Edoho, Udoinyang G Inyang, Ifiok J Udo, Itemobong S Ekaidem, Francis Osang, Nseobong P Uto, Joseph I Geoffery","doi":"10.2174/1570162X20666220210142209","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Effective global antiretroviral vaccines and therapeutic strategies depend on the diversity, evolution, and epidemiology of their various strains as well as their transmission and pathogenesis. Most viral disease-causing particles are clustered into a taxonomy of subtypes to suggest pointers toward nucleotide-specific vaccines or therapeutic applications of clinical significance sufficient for sequence-specific diagnosis and homologous viral studies. These are very useful to formulate predictors to induce cross-resistance to some retroviral control drugs being used across study areas.</p><p><strong>Objective: </strong>This research proposed a collaborative framework of hybridized (Machine Learning and Natural Language Processing) techniques to discover hidden genome patterns and feature predictors for HIV-1 genome sequences mining.</p><p><strong>Methods: </strong>630 human HIV-1 genome sequences above 8500 bps were excavated from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov) for 21 countries across different continents, except for Antarctica. These sequences were transformed and learned using a self-organizing map (SOM). To discriminate emerging/new sub-strain(s), the HIV-1 reference genome was included as part of the input isolates/samples during the training. After training the SOM, component planes defining pattern clusters of the input datasets were generated for cognitive knowledge mining and subsequent labeling of the datasets. Additional genome features, including dinucleotide transmission recurrences, codon recurrences, and mutation recurrences, were finally extracted from the raw genomes to construct output classification targets for supervised learning.</p><p><strong>Results: </strong>SOM training explains the inherent pattern diversity of HIV-1 genomes as well as interand intra-country transmissions in which mobility might play an active role, as corroborated by the literature. Nine sub-strains were discovered after disassembling the SOM correlation hunting matrix space attributed to disparate clusters. Cognitive knowledge mining separated similar pattern clusters bounded by a certain degree of correlation range, as discovered by the SOM. Kruskal-Wallis ranksum test and Wilcoxon rank-sum test showed statistically significant variations in dinucleotide, codon, and mutation patterns.</p><p><strong>Conclusion: </strong>Results of the discovered sub-strains and response clusters visualizations corroborate the existing literature, with significant haplotype variations. The proposed framework would assist in the development of decision support systems for easy contact tracing, infectious disease surveillance, and studying the progressive evolution of the reference HIV-1 genome.</p>","PeriodicalId":10911,"journal":{"name":"Current HIV Research","volume":"20 2","pages":"163-183"},"PeriodicalIF":0.8000,"publicationDate":"2022-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Collaborative Mining of Whole Genome Sequences for Intelligent HIV-1 Sub-Strain(s) Discovery.\",\"authors\":\"Moses E Ekpenyong, Anthony A Adegoke, Mercy E Edoho, Udoinyang G Inyang, Ifiok J Udo, Itemobong S Ekaidem, Francis Osang, Nseobong P Uto, Joseph I Geoffery\",\"doi\":\"10.2174/1570162X20666220210142209\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Effective global antiretroviral vaccines and therapeutic strategies depend on the diversity, evolution, and epidemiology of their various strains as well as their transmission and pathogenesis. Most viral disease-causing particles are clustered into a taxonomy of subtypes to suggest pointers toward nucleotide-specific vaccines or therapeutic applications of clinical significance sufficient for sequence-specific diagnosis and homologous viral studies. These are very useful to formulate predictors to induce cross-resistance to some retroviral control drugs being used across study areas.</p><p><strong>Objective: </strong>This research proposed a collaborative framework of hybridized (Machine Learning and Natural Language Processing) techniques to discover hidden genome patterns and feature predictors for HIV-1 genome sequences mining.</p><p><strong>Methods: </strong>630 human HIV-1 genome sequences above 8500 bps were excavated from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov) for 21 countries across different continents, except for Antarctica. These sequences were transformed and learned using a self-organizing map (SOM). To discriminate emerging/new sub-strain(s), the HIV-1 reference genome was included as part of the input isolates/samples during the training. After training the SOM, component planes defining pattern clusters of the input datasets were generated for cognitive knowledge mining and subsequent labeling of the datasets. Additional genome features, including dinucleotide transmission recurrences, codon recurrences, and mutation recurrences, were finally extracted from the raw genomes to construct output classification targets for supervised learning.</p><p><strong>Results: </strong>SOM training explains the inherent pattern diversity of HIV-1 genomes as well as interand intra-country transmissions in which mobility might play an active role, as corroborated by the literature. Nine sub-strains were discovered after disassembling the SOM correlation hunting matrix space attributed to disparate clusters. Cognitive knowledge mining separated similar pattern clusters bounded by a certain degree of correlation range, as discovered by the SOM. Kruskal-Wallis ranksum test and Wilcoxon rank-sum test showed statistically significant variations in dinucleotide, codon, and mutation patterns.</p><p><strong>Conclusion: </strong>Results of the discovered sub-strains and response clusters visualizations corroborate the existing literature, with significant haplotype variations. The proposed framework would assist in the development of decision support systems for easy contact tracing, infectious disease surveillance, and studying the progressive evolution of the reference HIV-1 genome.</p>\",\"PeriodicalId\":10911,\"journal\":{\"name\":\"Current HIV Research\",\"volume\":\"20 2\",\"pages\":\"163-183\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2022-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current HIV Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2174/1570162X20666220210142209\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current HIV Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2174/1570162X20666220210142209","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:有效的全球抗逆转录病毒疫苗和治疗策略取决于其各种菌株的多样性、进化和流行病学,以及它们的传播和发病机制。大多数病毒致病颗粒被聚集成一个亚型分类,以提示核苷酸特异性疫苗或具有临床意义的治疗应用,足以进行序列特异性诊断和同源病毒研究。这对于制定预测因子以诱导对跨研究区域使用的某些抗逆转录病毒控制药物产生交叉耐药性非常有用。目的:本研究提出了一个混合(机器学习和自然语言处理)技术的协作框架,以发现隐藏的基因组模式和特征预测因子,用于HIV-1基因组序列挖掘。方法:从美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)数据库(https://www.ncbi.nlm.nih.gov)中提取除南极洲以外的21个国家的630条8500 bps以上的人类HIV-1基因组序列。使用自组织映射(SOM)对这些序列进行转换和学习。为了区分新出现的/新的亚毒株,HIV-1参考基因组在训练过程中被作为输入分离株/样本的一部分。SOM训练完成后,生成定义输入数据集模式聚类的组件平面,用于认知知识挖掘和随后的数据集标注。最后,从原始基因组中提取额外的基因组特征,包括二核苷酸传递递归、密码子递归和突变递归,以构建监督学习的输出分类目标。结果:SOM培训解释了HIV-1基因组的固有模式多样性,以及国家间和国家内传播,其中流动性可能发挥积极作用,正如文献所证实的那样。通过对隶属于不同簇的SOM相关狩猎矩阵空间进行分解,发现了9个子菌株。认知知识挖掘在一定程度的关联范围内分离相似的模式簇,这是SOM发现的。Kruskal-Wallis秩和检验和Wilcoxon秩和检验显示,二核苷酸、密码子和突变模式的差异具有统计学意义。结论:发现的亚菌株和反应簇的可视化结果与已有文献一致,具有明显的单倍型变异。拟议的框架将有助于开发决策支持系统,以便于接触者追踪、传染病监测和研究参考HIV-1基因组的渐进进化。
Collaborative Mining of Whole Genome Sequences for Intelligent HIV-1 Sub-Strain(s) Discovery.
Background: Effective global antiretroviral vaccines and therapeutic strategies depend on the diversity, evolution, and epidemiology of their various strains as well as their transmission and pathogenesis. Most viral disease-causing particles are clustered into a taxonomy of subtypes to suggest pointers toward nucleotide-specific vaccines or therapeutic applications of clinical significance sufficient for sequence-specific diagnosis and homologous viral studies. These are very useful to formulate predictors to induce cross-resistance to some retroviral control drugs being used across study areas.
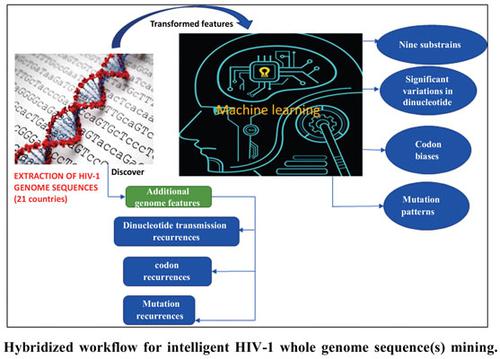
Objective: This research proposed a collaborative framework of hybridized (Machine Learning and Natural Language Processing) techniques to discover hidden genome patterns and feature predictors for HIV-1 genome sequences mining.
Methods: 630 human HIV-1 genome sequences above 8500 bps were excavated from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov) for 21 countries across different continents, except for Antarctica. These sequences were transformed and learned using a self-organizing map (SOM). To discriminate emerging/new sub-strain(s), the HIV-1 reference genome was included as part of the input isolates/samples during the training. After training the SOM, component planes defining pattern clusters of the input datasets were generated for cognitive knowledge mining and subsequent labeling of the datasets. Additional genome features, including dinucleotide transmission recurrences, codon recurrences, and mutation recurrences, were finally extracted from the raw genomes to construct output classification targets for supervised learning.
Results: SOM training explains the inherent pattern diversity of HIV-1 genomes as well as interand intra-country transmissions in which mobility might play an active role, as corroborated by the literature. Nine sub-strains were discovered after disassembling the SOM correlation hunting matrix space attributed to disparate clusters. Cognitive knowledge mining separated similar pattern clusters bounded by a certain degree of correlation range, as discovered by the SOM. Kruskal-Wallis ranksum test and Wilcoxon rank-sum test showed statistically significant variations in dinucleotide, codon, and mutation patterns.
Conclusion: Results of the discovered sub-strains and response clusters visualizations corroborate the existing literature, with significant haplotype variations. The proposed framework would assist in the development of decision support systems for easy contact tracing, infectious disease surveillance, and studying the progressive evolution of the reference HIV-1 genome.
期刊介绍:
Current HIV Research covers all the latest and outstanding developments of HIV research by publishing original research, review articles and guest edited thematic issues. The novel pioneering work in the basic and clinical fields on all areas of HIV research covers: virus replication and gene expression, HIV assembly, virus-cell interaction, viral pathogenesis, epidemiology and transmission, anti-retroviral therapy and adherence, drug discovery, the latest developments in HIV/AIDS vaccines and animal models, mechanisms and interactions with AIDS related diseases, social and public health issues related to HIV disease, and prevention of viral infection. Periodically, the journal invites guest editors to devote an issue on a particular area of HIV research of great interest that increases our understanding of the virus and its complex interaction with the host.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


