Mingliang Li, He Huang, Chunlian Ke, Lei Tan, Jiezhong Wu, Shilei Xu, Xusheng Tu
{"title":"基于加权基因共表达网络分析和支持向量机算法的脓毒症患者四基因诊断特征识别","authors":"Mingliang Li, He Huang, Chunlian Ke, Lei Tan, Jiezhong Wu, Shilei Xu, Xusheng Tu","doi":"10.1186/s41065-021-00215-8","DOIUrl":null,"url":null,"abstract":"<p><p>Sepsis is a life-threatening condition in which the immune response is directed towards the host tissues, causing organ failure. Since sepsis does not present with specific symptoms, its diagnosis is often delayed. The lack of diagnostic accuracy results in a non-specific diagnosis, and to date, a standard diagnostic test to detect sepsis in patients remains lacking. Therefore, it is vital to identify sepsis-related diagnostic genes. This study aimed to conduct an integrated analysis to assess the immune scores of samples from patients diagnosed with sepsis and normal samples, followed by weighted gene co-expression network analysis (WGCNA) to identify immune infiltration-related genes and potential transcriptome markers in sepsis. Furthermore, gene regulatory networks were established to screen diagnostic markers for sepsis based on the protein-protein interaction networks involving these immune infiltration-related genes. Moreover, we integrated WGCNA with the support vector machine (SVM) algorithm to build a diagnostic model for sepsis. Results showed that the immune score was significantly lower in the samples from patients with sepsis than in normal samples. A total of 328 and 333 genes were positively and negatively correlated with the immune score, respectively. Using the MCODE plugin in Cytoscape, we identified four modules, and through functional annotation, we found that these modules were related to the immune response. Gene Ontology functional enrichment analysis showed that the identified genes were associated with functions such as neutrophil degranulation, neutrophil activation in the immune response, neutrophil activation, and neutrophil-mediated immunity. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed the enrichment of pathways such as primary immunodeficiency, Th1- and Th2-cell differentiation, T-cell receptor signaling pathway, and natural killer cell-mediated cytotoxicity. Finally, we identified a four-gene signature, containing the hub genes LCK, CCL5, ITGAM, and MMP9, and established a model that could be used to diagnose patients with sepsis.</p>","PeriodicalId":12862,"journal":{"name":"Hereditas","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2022-02-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8859894/pdf/","citationCount":"3","resultStr":"{\"title\":\"Identification of a novel four-gene diagnostic signature for patients with sepsis by integrating weighted gene co-expression network analysis and support vector machine algorithm.\",\"authors\":\"Mingliang Li, He Huang, Chunlian Ke, Lei Tan, Jiezhong Wu, Shilei Xu, Xusheng Tu\",\"doi\":\"10.1186/s41065-021-00215-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Sepsis is a life-threatening condition in which the immune response is directed towards the host tissues, causing organ failure. Since sepsis does not present with specific symptoms, its diagnosis is often delayed. The lack of diagnostic accuracy results in a non-specific diagnosis, and to date, a standard diagnostic test to detect sepsis in patients remains lacking. Therefore, it is vital to identify sepsis-related diagnostic genes. This study aimed to conduct an integrated analysis to assess the immune scores of samples from patients diagnosed with sepsis and normal samples, followed by weighted gene co-expression network analysis (WGCNA) to identify immune infiltration-related genes and potential transcriptome markers in sepsis. Furthermore, gene regulatory networks were established to screen diagnostic markers for sepsis based on the protein-protein interaction networks involving these immune infiltration-related genes. Moreover, we integrated WGCNA with the support vector machine (SVM) algorithm to build a diagnostic model for sepsis. Results showed that the immune score was significantly lower in the samples from patients with sepsis than in normal samples. A total of 328 and 333 genes were positively and negatively correlated with the immune score, respectively. Using the MCODE plugin in Cytoscape, we identified four modules, and through functional annotation, we found that these modules were related to the immune response. Gene Ontology functional enrichment analysis showed that the identified genes were associated with functions such as neutrophil degranulation, neutrophil activation in the immune response, neutrophil activation, and neutrophil-mediated immunity. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed the enrichment of pathways such as primary immunodeficiency, Th1- and Th2-cell differentiation, T-cell receptor signaling pathway, and natural killer cell-mediated cytotoxicity. Finally, we identified a four-gene signature, containing the hub genes LCK, CCL5, ITGAM, and MMP9, and established a model that could be used to diagnose patients with sepsis.</p>\",\"PeriodicalId\":12862,\"journal\":{\"name\":\"Hereditas\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2022-02-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8859894/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hereditas\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s41065-021-00215-8\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-021-00215-8","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Identification of a novel four-gene diagnostic signature for patients with sepsis by integrating weighted gene co-expression network analysis and support vector machine algorithm.
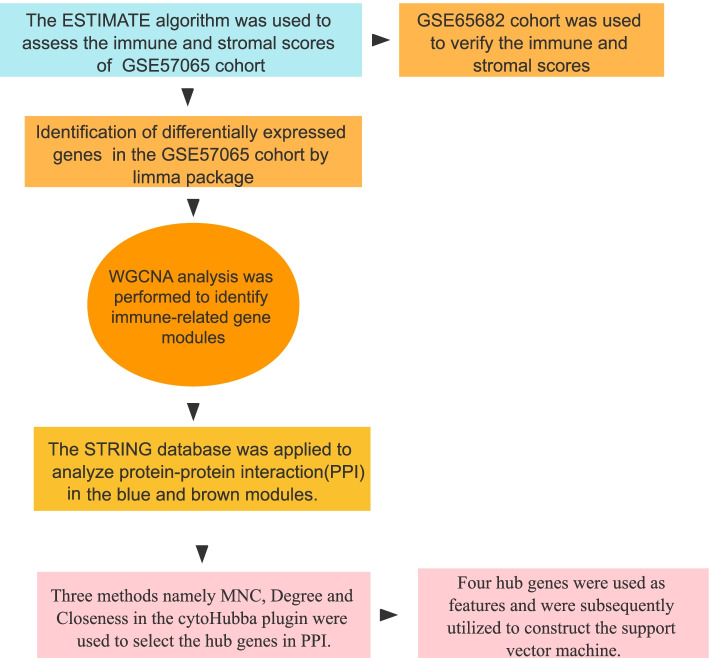
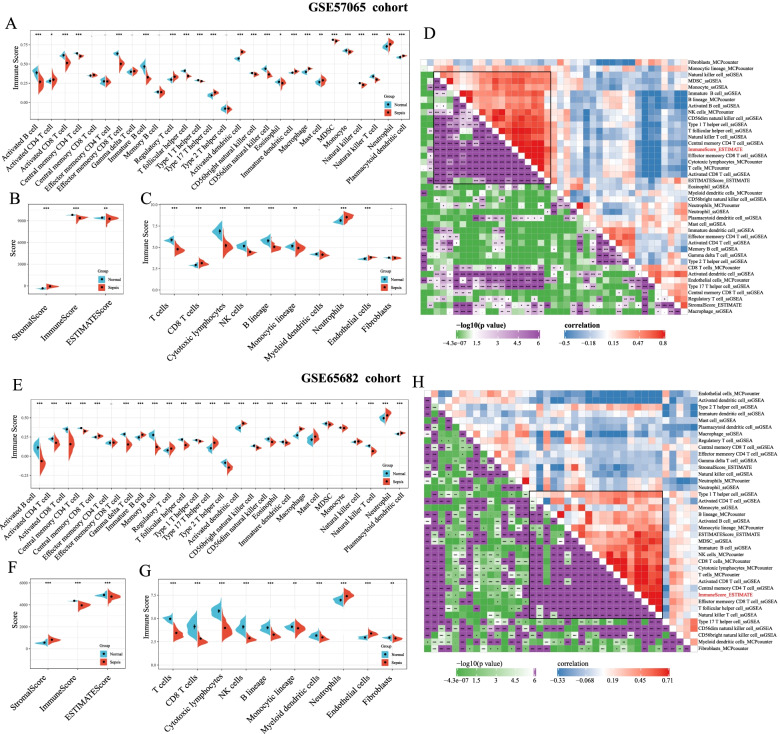
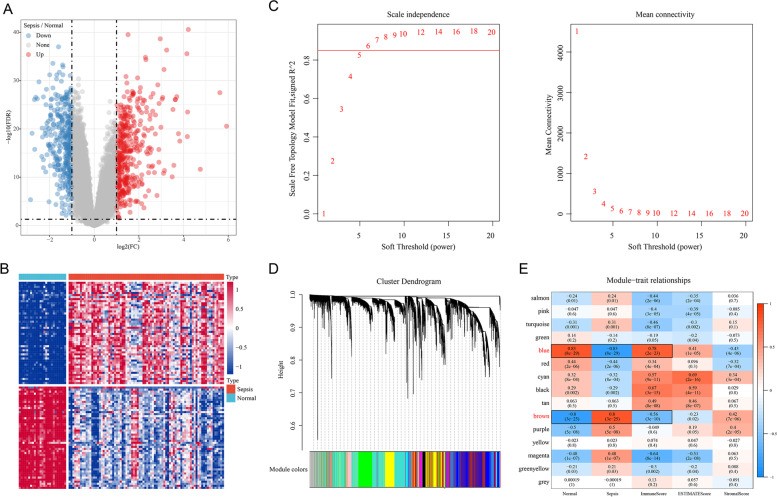
Sepsis is a life-threatening condition in which the immune response is directed towards the host tissues, causing organ failure. Since sepsis does not present with specific symptoms, its diagnosis is often delayed. The lack of diagnostic accuracy results in a non-specific diagnosis, and to date, a standard diagnostic test to detect sepsis in patients remains lacking. Therefore, it is vital to identify sepsis-related diagnostic genes. This study aimed to conduct an integrated analysis to assess the immune scores of samples from patients diagnosed with sepsis and normal samples, followed by weighted gene co-expression network analysis (WGCNA) to identify immune infiltration-related genes and potential transcriptome markers in sepsis. Furthermore, gene regulatory networks were established to screen diagnostic markers for sepsis based on the protein-protein interaction networks involving these immune infiltration-related genes. Moreover, we integrated WGCNA with the support vector machine (SVM) algorithm to build a diagnostic model for sepsis. Results showed that the immune score was significantly lower in the samples from patients with sepsis than in normal samples. A total of 328 and 333 genes were positively and negatively correlated with the immune score, respectively. Using the MCODE plugin in Cytoscape, we identified four modules, and through functional annotation, we found that these modules were related to the immune response. Gene Ontology functional enrichment analysis showed that the identified genes were associated with functions such as neutrophil degranulation, neutrophil activation in the immune response, neutrophil activation, and neutrophil-mediated immunity. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed the enrichment of pathways such as primary immunodeficiency, Th1- and Th2-cell differentiation, T-cell receptor signaling pathway, and natural killer cell-mediated cytotoxicity. Finally, we identified a four-gene signature, containing the hub genes LCK, CCL5, ITGAM, and MMP9, and established a model that could be used to diagnose patients with sepsis.
HereditasBiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
3.80
自引率
3.70%
发文量
0
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


