{"title":"两个早花樱桃(Cerasus × kanzakura)品种‘Kawazu-zakura’和‘Atami-zakura’的基因组测序和分析。","authors":"Kenta Shirasawa, Akihiro Itai, Sachiko Isobe","doi":"10.1093/dnares/dsab026","DOIUrl":null,"url":null,"abstract":"<p><p>To gain genetic insights into the early-flowering phenotype of ornamental cherry, also known as sakura, we determined the genome sequences of two early-flowering cherry (Cerasus × kanzakura) varieties, 'Kawazu-zakura' and 'Atami-zakura'. Because the two varieties are interspecific hybrids, likely derived from crosses between Cerasus campanulata (early-flowering species) and Cerasus speciosa, we employed the haplotype-resolved sequence assembly strategy. Genome sequence reads obtained from each variety by single-molecule real-time sequencing (SMRT) were split into two subsets, based on the genome sequence information of the two probable ancestors, and assembled to obtain haplotype-phased genome sequences. The resultant genome assembly of 'Kawazu-zakura' spanned 519.8 Mb with 1,544 contigs and an N50 value of 1,220.5 kb, while that of 'Atami-zakura' totalled 509.6 Mb with 2,180 contigs and an N50 value of 709.1 kb. A total of 72,702 and 69,528 potential protein-coding genes were predicted in the genome assemblies of 'Kawazu-zakura' and 'Atami-zakura', respectively. Gene clustering analysis identified 2,634 clusters uniquely presented in the C. campanulata haplotype sequences, which might contribute to its early-flowering phenotype. Genome sequences determined in this study provide fundamental information for elucidating the molecular and genetic mechanisms underlying the early-flowering phenotype of ornamental cherry tree varieties and their relatives.</p>","PeriodicalId":51014,"journal":{"name":"DNA Research","volume":"28 6","pages":""},"PeriodicalIF":3.9000,"publicationDate":"2021-10-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8643691/pdf/","citationCount":"6","resultStr":"{\"title\":\"Genome sequencing and analysis of two early-flowering cherry (Cerasus × kanzakura) varieties, 'Kawazu-zakura' and 'Atami-zakura'.\",\"authors\":\"Kenta Shirasawa, Akihiro Itai, Sachiko Isobe\",\"doi\":\"10.1093/dnares/dsab026\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>To gain genetic insights into the early-flowering phenotype of ornamental cherry, also known as sakura, we determined the genome sequences of two early-flowering cherry (Cerasus × kanzakura) varieties, 'Kawazu-zakura' and 'Atami-zakura'. Because the two varieties are interspecific hybrids, likely derived from crosses between Cerasus campanulata (early-flowering species) and Cerasus speciosa, we employed the haplotype-resolved sequence assembly strategy. Genome sequence reads obtained from each variety by single-molecule real-time sequencing (SMRT) were split into two subsets, based on the genome sequence information of the two probable ancestors, and assembled to obtain haplotype-phased genome sequences. The resultant genome assembly of 'Kawazu-zakura' spanned 519.8 Mb with 1,544 contigs and an N50 value of 1,220.5 kb, while that of 'Atami-zakura' totalled 509.6 Mb with 2,180 contigs and an N50 value of 709.1 kb. A total of 72,702 and 69,528 potential protein-coding genes were predicted in the genome assemblies of 'Kawazu-zakura' and 'Atami-zakura', respectively. Gene clustering analysis identified 2,634 clusters uniquely presented in the C. campanulata haplotype sequences, which might contribute to its early-flowering phenotype. Genome sequences determined in this study provide fundamental information for elucidating the molecular and genetic mechanisms underlying the early-flowering phenotype of ornamental cherry tree varieties and their relatives.</p>\",\"PeriodicalId\":51014,\"journal\":{\"name\":\"DNA Research\",\"volume\":\"28 6\",\"pages\":\"\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2021-10-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8643691/pdf/\",\"citationCount\":\"6\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"DNA Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/dnares/dsab026\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"DNA Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/dnares/dsab026","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genome sequencing and analysis of two early-flowering cherry (Cerasus × kanzakura) varieties, 'Kawazu-zakura' and 'Atami-zakura'.
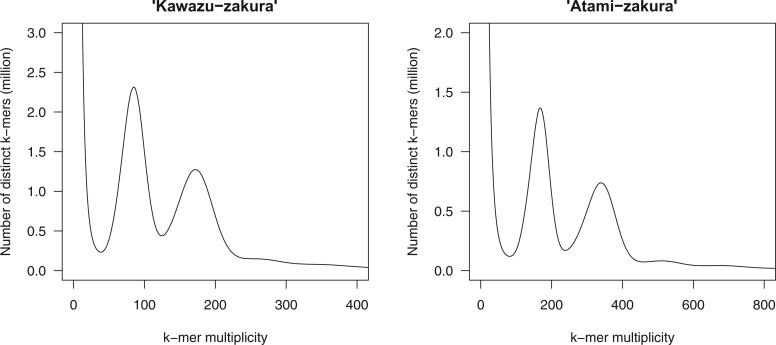
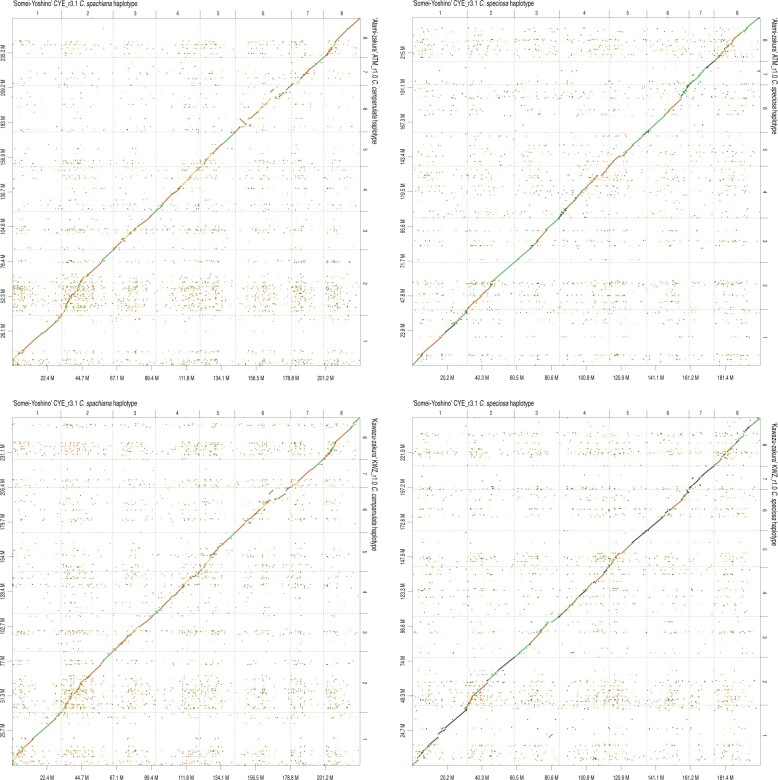
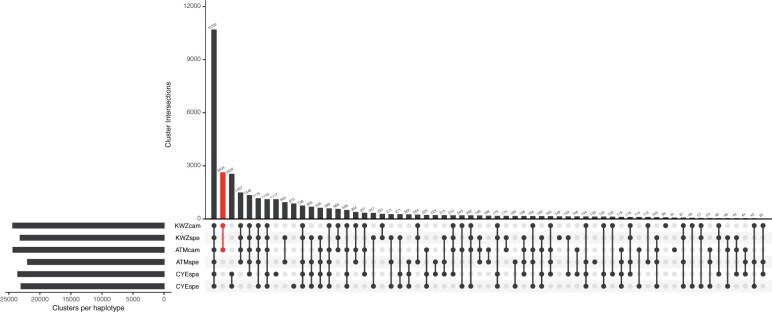
To gain genetic insights into the early-flowering phenotype of ornamental cherry, also known as sakura, we determined the genome sequences of two early-flowering cherry (Cerasus × kanzakura) varieties, 'Kawazu-zakura' and 'Atami-zakura'. Because the two varieties are interspecific hybrids, likely derived from crosses between Cerasus campanulata (early-flowering species) and Cerasus speciosa, we employed the haplotype-resolved sequence assembly strategy. Genome sequence reads obtained from each variety by single-molecule real-time sequencing (SMRT) were split into two subsets, based on the genome sequence information of the two probable ancestors, and assembled to obtain haplotype-phased genome sequences. The resultant genome assembly of 'Kawazu-zakura' spanned 519.8 Mb with 1,544 contigs and an N50 value of 1,220.5 kb, while that of 'Atami-zakura' totalled 509.6 Mb with 2,180 contigs and an N50 value of 709.1 kb. A total of 72,702 and 69,528 potential protein-coding genes were predicted in the genome assemblies of 'Kawazu-zakura' and 'Atami-zakura', respectively. Gene clustering analysis identified 2,634 clusters uniquely presented in the C. campanulata haplotype sequences, which might contribute to its early-flowering phenotype. Genome sequences determined in this study provide fundamental information for elucidating the molecular and genetic mechanisms underlying the early-flowering phenotype of ornamental cherry tree varieties and their relatives.
期刊介绍:
DNA Research is an internationally peer-reviewed journal which aims at publishing papers of highest quality in broad aspects of DNA and genome-related research. Emphasis will be made on the following subjects: 1) Sequencing and characterization of genomes/important genomic regions, 2) Comprehensive analysis of the functions of genes, gene families and genomes, 3) Techniques and equipments useful for structural and functional analysis of genes, gene families and genomes, 4) Computer algorithms and/or their applications relevant to structural and functional analysis of genes and genomes. The journal also welcomes novel findings in other scientific disciplines related to genomes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


