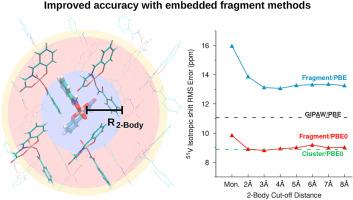
{"title":"分子晶体中精确的基于片段的51-V化学位移预测","authors":"Amanda Mathews, Joshua D. Hartman","doi":"10.1016/j.ssnmr.2021.101733","DOIUrl":null,"url":null,"abstract":"<div><p><span>Nuclear magnetic resonance (NMR) spectroscopy plays a crucial role in determining molecular structure for complex biological and pharmaceutical compounds. NMR investigations are increasingly reliant on computation for mapping spectral features to chemical structures. Here we benchmark the accuracy of fragment-based </span><sup>51</sup><span>V chemical shielding tensor calculations using a training set comprised of 10 biologically and pharmaceutically relevant oxovanadium complexes. Using our self-consistent reproduction of the Madelung potential (SCRMP) electrostatic embedding model, we demonstrate comparable performance between fragment methods and computationally demanding cluster-based techniques. Specifically, fragment methods employing hybrid density functionals are capable of reproducing the experimental </span><sup>51</sup>V isotropic chemical shifts with a training set rms error of <span><math><mrow><mo>~</mo></mrow></math></span><span>9 ppm, representing a 20% improvement over traditional plane wave techniques. We provide training set-derived linear regression models for mapping the absolute shieldings obtained from computation to the experimentally determined chemical shifts using four common density functionals; PBE0, B3LYP, PBE, and BLYP. Finally, we establish the utility of fragment methods and the reported regression parameters examining four oxovanadium structures excluded from the training set including the tetracoordinate oxovanadium silicate </span><span><math><mrow><msub><mrow><mo>(</mo><mrow><mi>P</mi><msub><mi>h</mi><mn>3</mn></msub><mi>S</mi><mi>i</mi><mi>O</mi></mrow><mo>)</mo></mrow><mn>3</mn></msub><mi>V</mi><mi>O</mi></mrow></math></span>, VO<sup>15</sup>NGlySalbz which contains redox-active ligands, and the solid-state form of the common <sup>51</sup>V NMR reference compound <span><math><mrow><mi>V</mi><mi>O</mi><mi>C</mi><msub><mi>l</mi><mn>3</mn></msub></mrow></math></span>.</p></div>","PeriodicalId":21937,"journal":{"name":"Solid state nuclear magnetic resonance","volume":"114 ","pages":"Article 101733"},"PeriodicalIF":2.4000,"publicationDate":"2021-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.ssnmr.2021.101733","citationCount":"2","resultStr":"{\"title\":\"Accurate fragment-based 51-V chemical shift predictions in molecular crystals\",\"authors\":\"Amanda Mathews, Joshua D. Hartman\",\"doi\":\"10.1016/j.ssnmr.2021.101733\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><span>Nuclear magnetic resonance (NMR) spectroscopy plays a crucial role in determining molecular structure for complex biological and pharmaceutical compounds. NMR investigations are increasingly reliant on computation for mapping spectral features to chemical structures. Here we benchmark the accuracy of fragment-based </span><sup>51</sup><span>V chemical shielding tensor calculations using a training set comprised of 10 biologically and pharmaceutically relevant oxovanadium complexes. Using our self-consistent reproduction of the Madelung potential (SCRMP) electrostatic embedding model, we demonstrate comparable performance between fragment methods and computationally demanding cluster-based techniques. Specifically, fragment methods employing hybrid density functionals are capable of reproducing the experimental </span><sup>51</sup>V isotropic chemical shifts with a training set rms error of <span><math><mrow><mo>~</mo></mrow></math></span><span>9 ppm, representing a 20% improvement over traditional plane wave techniques. We provide training set-derived linear regression models for mapping the absolute shieldings obtained from computation to the experimentally determined chemical shifts using four common density functionals; PBE0, B3LYP, PBE, and BLYP. Finally, we establish the utility of fragment methods and the reported regression parameters examining four oxovanadium structures excluded from the training set including the tetracoordinate oxovanadium silicate </span><span><math><mrow><msub><mrow><mo>(</mo><mrow><mi>P</mi><msub><mi>h</mi><mn>3</mn></msub><mi>S</mi><mi>i</mi><mi>O</mi></mrow><mo>)</mo></mrow><mn>3</mn></msub><mi>V</mi><mi>O</mi></mrow></math></span>, VO<sup>15</sup>NGlySalbz which contains redox-active ligands, and the solid-state form of the common <sup>51</sup>V NMR reference compound <span><math><mrow><mi>V</mi><mi>O</mi><mi>C</mi><msub><mi>l</mi><mn>3</mn></msub></mrow></math></span>.</p></div>\",\"PeriodicalId\":21937,\"journal\":{\"name\":\"Solid state nuclear magnetic resonance\",\"volume\":\"114 \",\"pages\":\"Article 101733\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2021-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1016/j.ssnmr.2021.101733\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Solid state nuclear magnetic resonance\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0926204021000217\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Solid state nuclear magnetic resonance","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0926204021000217","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Accurate fragment-based 51-V chemical shift predictions in molecular crystals
Nuclear magnetic resonance (NMR) spectroscopy plays a crucial role in determining molecular structure for complex biological and pharmaceutical compounds. NMR investigations are increasingly reliant on computation for mapping spectral features to chemical structures. Here we benchmark the accuracy of fragment-based 51V chemical shielding tensor calculations using a training set comprised of 10 biologically and pharmaceutically relevant oxovanadium complexes. Using our self-consistent reproduction of the Madelung potential (SCRMP) electrostatic embedding model, we demonstrate comparable performance between fragment methods and computationally demanding cluster-based techniques. Specifically, fragment methods employing hybrid density functionals are capable of reproducing the experimental 51V isotropic chemical shifts with a training set rms error of 9 ppm, representing a 20% improvement over traditional plane wave techniques. We provide training set-derived linear regression models for mapping the absolute shieldings obtained from computation to the experimentally determined chemical shifts using four common density functionals; PBE0, B3LYP, PBE, and BLYP. Finally, we establish the utility of fragment methods and the reported regression parameters examining four oxovanadium structures excluded from the training set including the tetracoordinate oxovanadium silicate , VO15NGlySalbz which contains redox-active ligands, and the solid-state form of the common 51V NMR reference compound .
期刊介绍:
The journal Solid State Nuclear Magnetic Resonance publishes original manuscripts of high scientific quality dealing with all experimental and theoretical aspects of solid state NMR. This includes advances in instrumentation, development of new experimental techniques and methodology, new theoretical insights, new data processing and simulation methods, and original applications of established or novel methods to scientific problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


