{"title":"以横纹肌溶解为表现的超长链酰基辅酶A脱氢酶缺乏:斯里兰卡首例报告。","authors":"Maheshi Wijayabandara, Champika Gamakaranage, Dineshani Hettiarachchi","doi":"10.1155/2020/8894518","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Rhabdomyolysis can be either inherited or acquired such as in metabolic myopathies. Very-long-chain acyl-CoA dehydrogenase deficiency is a rare fatty acid oxidation disorder which presents with different phenotypes, and the mild adult form can present as intermittent rhabdomyolysis. Here, we present the first adult case of very-long-chain acyl-CoA dehydrogenase deficiency presenting as rhabdomyolysis in a Sri Lankan patient. <i>Case Presentation</i>. A 36-year-old Sri Lankan man who was born to consanguineous parents presented with severe generalized muscle pain, stiffness, and dark-coloured urine for three days following prolonged low-intensity activity. Since fourteen years of age, he has had multiple similar episodes, where one episode was complicated with acute kidney injury. His eldest brother also suffered from the similar episode. Examination revealed only generalized muscle tenderness without any weakness. His creatine phosphokinase level was above 50,000 IU/L, and he had myoglobinuria. Molecular genetic tests confirmed the diagnosis of very-long-chain acyl-CoA dehydrogenase deficiency. Following a successful recovery devoid of complications, he remained asymptomatic with lifestyle adjustments.</p><p><strong>Conclusion: </strong>Very-long-chain acyl-CoA dehydrogenase deficiency is a rare inherited cause of metabolic myopathy that gives rise to intermittent rhabdomyolysis in adults. Prompt diagnosis is essential to prevent complications and prevent its recurrence.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2020 ","pages":"8894518"},"PeriodicalIF":0.0000,"publicationDate":"2020-10-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2020/8894518","citationCount":"1","resultStr":"{\"title\":\"Very-Long-Chain Acyl-Co-Enzyme A Dehydrogenase Deficiency Presenting as Rhabdomyolysis: First Case Report from Sri Lanka.\",\"authors\":\"Maheshi Wijayabandara, Champika Gamakaranage, Dineshani Hettiarachchi\",\"doi\":\"10.1155/2020/8894518\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Rhabdomyolysis can be either inherited or acquired such as in metabolic myopathies. Very-long-chain acyl-CoA dehydrogenase deficiency is a rare fatty acid oxidation disorder which presents with different phenotypes, and the mild adult form can present as intermittent rhabdomyolysis. Here, we present the first adult case of very-long-chain acyl-CoA dehydrogenase deficiency presenting as rhabdomyolysis in a Sri Lankan patient. <i>Case Presentation</i>. A 36-year-old Sri Lankan man who was born to consanguineous parents presented with severe generalized muscle pain, stiffness, and dark-coloured urine for three days following prolonged low-intensity activity. Since fourteen years of age, he has had multiple similar episodes, where one episode was complicated with acute kidney injury. His eldest brother also suffered from the similar episode. Examination revealed only generalized muscle tenderness without any weakness. His creatine phosphokinase level was above 50,000 IU/L, and he had myoglobinuria. Molecular genetic tests confirmed the diagnosis of very-long-chain acyl-CoA dehydrogenase deficiency. Following a successful recovery devoid of complications, he remained asymptomatic with lifestyle adjustments.</p><p><strong>Conclusion: </strong>Very-long-chain acyl-CoA dehydrogenase deficiency is a rare inherited cause of metabolic myopathy that gives rise to intermittent rhabdomyolysis in adults. Prompt diagnosis is essential to prevent complications and prevent its recurrence.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2020 \",\"pages\":\"8894518\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-10-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2020/8894518\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2020/8894518\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2020/8894518","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Very-Long-Chain Acyl-Co-Enzyme A Dehydrogenase Deficiency Presenting as Rhabdomyolysis: First Case Report from Sri Lanka.
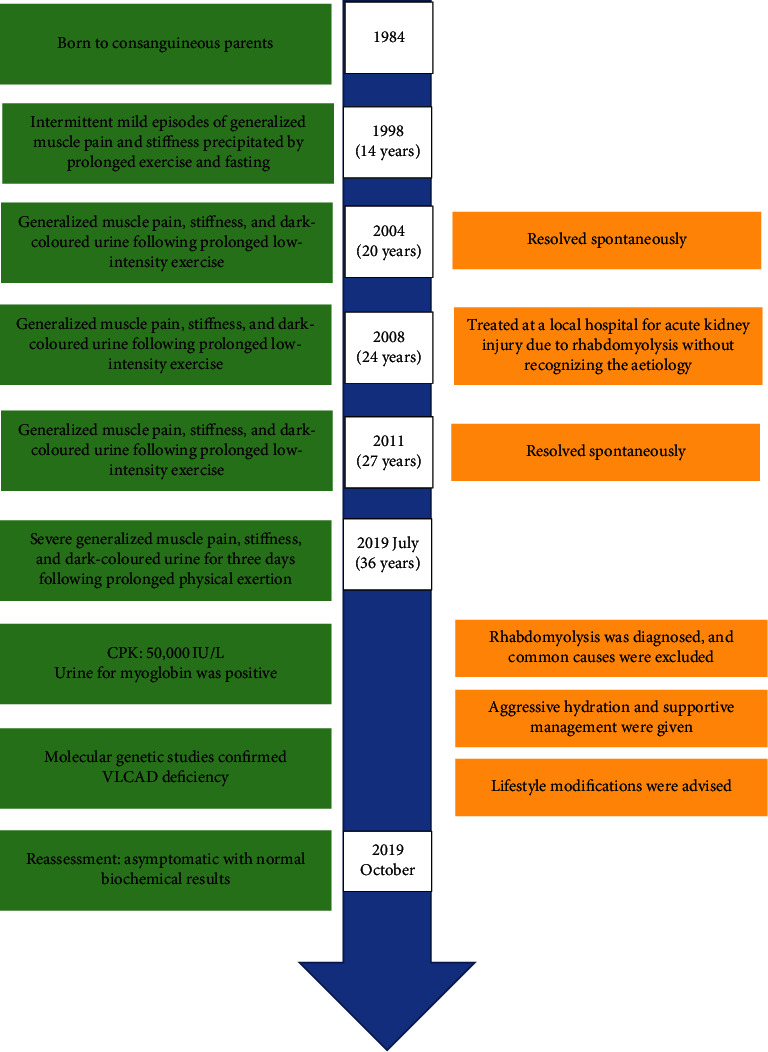
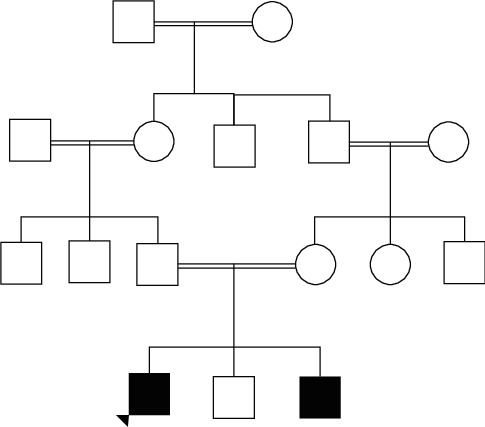
Background: Rhabdomyolysis can be either inherited or acquired such as in metabolic myopathies. Very-long-chain acyl-CoA dehydrogenase deficiency is a rare fatty acid oxidation disorder which presents with different phenotypes, and the mild adult form can present as intermittent rhabdomyolysis. Here, we present the first adult case of very-long-chain acyl-CoA dehydrogenase deficiency presenting as rhabdomyolysis in a Sri Lankan patient. Case Presentation. A 36-year-old Sri Lankan man who was born to consanguineous parents presented with severe generalized muscle pain, stiffness, and dark-coloured urine for three days following prolonged low-intensity activity. Since fourteen years of age, he has had multiple similar episodes, where one episode was complicated with acute kidney injury. His eldest brother also suffered from the similar episode. Examination revealed only generalized muscle tenderness without any weakness. His creatine phosphokinase level was above 50,000 IU/L, and he had myoglobinuria. Molecular genetic tests confirmed the diagnosis of very-long-chain acyl-CoA dehydrogenase deficiency. Following a successful recovery devoid of complications, he remained asymptomatic with lifestyle adjustments.
Conclusion: Very-long-chain acyl-CoA dehydrogenase deficiency is a rare inherited cause of metabolic myopathy that gives rise to intermittent rhabdomyolysis in adults. Prompt diagnosis is essential to prevent complications and prevent its recurrence.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


